
Ликсумия 20 мкг инструкция, аналоги и состав
| Показания: | Для лікування дорослих на цукровий діабет 2 типу з метою досягнення глікемічного контролю в комбінації з пероральними протидіабетичними засобами та/або базальним інсуліном, коли ці препарати разом з дієтою та фізичними вправами не забезпечують належний контроль глікемії. |
| Форма випуска: | Розчин для ін'єкцій, 0,1 мг/мл по 3 мл (14 доз) у катриджі, вмонтованому у шприц-ручку № 1 |
| Производитель, страна: | Санофі-Авентіс Дойчланд ГмбХ, Німеччина |
| Действующее вещества: | 1 мл розчину містить: ліксисенатиду 0,1 мг;1 доза містить: ліксисенатиду 20 мкг |
| МНН: | Lixisenatide - Ликсисенатид |
| Регистрация: | UA/13440/01/02з 30.01.2014 по 30.01.2019. Приказ 432 від 15.07.2015 |
| Код АТХ: |
Склад:
діюча речовина:ліксисенатид;
1 мл розчину містить: ліксисенатиду 0,1 мг; 1 доза містить: ліксисенатиду 20 мкг;
допоміжні речовини: гліцерин (85%), натрію ацетат тригідрат, метіонін, м-крезол, кислота хлористоводнева концентрована, натрію гідроксид, вода для ін’єкцій.
Лікарська форма. Розчин для ін’єкцій.
Фармакотерапевтична група. Антидіабетичні препарати. Гіпоглікемізуючі препарати, за виключенням інсулінів. Код АТС А10В Х10.
Клінічні характеристики.
Показання Для лікування дорослих на цукровий діабет 2 типу з метою досягнення глікемічного контролю в комбінації з пероральними протидіабетичними засобами та/або базальним інсуліном, коли ці препарати разом з дієтою та фізичними вправами не забезпечують належний контроль глікемії.
Протипоказання. Гіперчутливість до діючої речовини або будь-якої допоміжної речовини.
Спосіб застосування та дози. Препарат призначає лікар. Для введення початкової дози препарату Ліксумія®, яка становить 10 мкг один раз на добу протягом 14 діб, рекомендовано застосовувати шприц-ручку Ліксумія® 10 мкг. Підтримуюча доза: прийом фіксованої підтримуючої дози препарату Ліксумія® − 20 мкг один раз на добу − починають на 15-й день лікування. Для введення підтримуючої дози препарат Ліксумія® доступний у розчині для ін’єкцій по 20 мкг.
Препарат Ліксумія® застосовують один раз на добу за одну годину до сніданку або до вечері. Якщо введення дози було пропущене, препарат слід ввести за годину до наступного прийому їжі.
При додаванні препарату Ліксумія® до терапії метформіном поточну дозу метформіну залишають незмінною.
При додаванні препарату Ліксумія® до терапії похідними сульфонілсечовини або базального інсуліну для зниження ризику гіпоглікемії слід розглянути доцільність зниження дози похідних сульфонілсечовини та базального інсуліну (див. розділ «Особливості застосування»). Препарат Ліксумія® не повинен застосовуватися одночасно із похідними сульфонілсечовини та базальним інсуліном у зв’язку з високим ризиком гіпоглікемії.
Застосування препарату Ліксумія® не потребує особливого моніторингу рівня глюкози у крові. Однак при застосуванні препарату в комбінації з похідними сульфонілсечовини чи базальним інсуліном моніторинг або самостійний моніторинг рівня глюкози крові може стати необхідним для коригування доз похідних сульфонілсечовини та базального інсуліну.
Особливі популяції.
Пацієнти літнього віку (від 65 років). Необхідність в корекції дози відсутня (див розділ «Фармакокінетика»). Клінічний досвід застосування препарату у пацієнтів віком ≥75 років обмежений (див. розділи «Фармакодинаміка» та «Фармакокінетика»).
Ниркова недостатність. У пацієнтів з нирковою недостатністю легкого ступеня тяжкості (з кліренсом креатиніну 50-80 мл/хв) необхідність в корекції дози відсутня. Терапевтичний досвід застосування препарату у пацієнтів з помірною нирковою недостатністю (кліренс креатиніну: 30–50 мл/хв) обмежений, тому Ліксумія® має використовуватися з обережністю у цієї популяції пацієнтів.
Оскільки досвід застосування препарату Ліксумія® у пацієнтів з тяжкою нирковою недостатністю (з кліренсом креатиніну нижче 30 мл/хв) або термінальною стадією ниркової недостатності відсутній, застосування препарату у таких пацієнтів не рекомендоване (див. розділ «Фармакокінетика»)
Печінкова недостатність. Необхідність в корекції дози у пацієнтів з печінковою недостатністю відсутня (див. розділ «Фармакокінетика»).
Спосіб застосування. Препарат вводять підшкірно в стегно, живіт або у верхню частину руки. Препарат Ліксумія® не можна вводити внутрішньовенно або внутрішньом’язово.
Інструкція з використання шприц-ручки
Ліксумія® – це попередньо заповнена шприц-ручка для ін'єкцій.
- Перед початком використання шприц-ручки проконсультуйтеся зі своїм лікарем, як правильно виконувати ін'єкцію.
- Слід застосовувати лише одну дозу на день.
- Кожна шприц-ручка Ліксумія® містить 14 фіксованих доз. Немає необхідності відміряти кожну дозу окремо.
- Якщо Ви не в змозі повністю виконувати всі інструкції з використання шприц-ручки самостійно або не можете користуватися нею (наприклад, через проблеми з зором), використовуйте її лише зі сторонньою допомогою.
Схематичне зображення шприц-ручки Ліксумія®
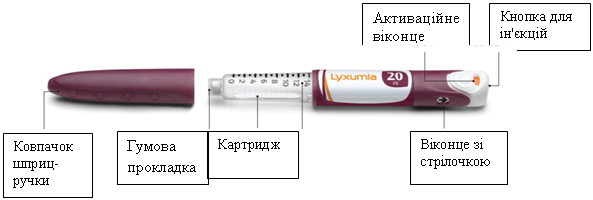


- Ця шприц-ручка призначена для застосування лише однією особою. Не дозволяється передавати її будь-кому іншому для застосування.
- Завжди перевіряйте етикетку, щоб переконатися, що Ви використовуєте саме потрібну шприц-ручку Ліксумія®. Крім того, переконайтеся, що не сплив термін придатності лікарського засобу. Застосування непридатних ліків може бути небезпечним для Вашого здоров'я.
- Не намагайтеся дістати рідину з картриджа за допомогою звичайного шприца.
Схематичне зображення голки (постачається окремо)
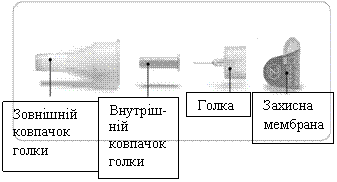
- Слід застосовувати лише голки, які призначені для використання з шприц-ручкою Ліксумія®. З шприц-ручкою Ліксумія® дозволяється використовувати одноразові голки від 29 до 32 калібрів. Проконсультуйтеся з лікуючим лікарем, фармацевтом або медсестрою, щоб підібрати калібр та довжину голки, які найкраще підходять саме Вам.
- Якщо ін'єкцію виконує інша особа, вона має бути обережною, щоб випадково не поранити когось голкою. Це може призвести до можливого перенесення інфекції.
- Для кожної ін'єкції завжди слід застосовувати нову голку. Це допомагає уникнути контамінації препарату Ліксумія® або можливого закупорювання голки.
ПОЧАТОК РОБОТИ
- Активуйте шприц-ручку у день першого її застосування.
- Перед введенням дози – перед виконанням ін'єкціїперш за все слід видалити надлишок рідини з нової шприц-ручки. Ця процедура виконується один раз і називається процесом «активації». Щоб активувати шприц-ручку, виконайте кроки 1-5, описані нижче.
- Активацію виконують для того, щоб переконатися, що шприц-ручка працює коректно і що доза для першої ін'єкції є правильною.
- Не слід виконувати процес активації повторно, тому що в такому випадку Ви вже не зможете отримати 14 доз лікарського засобу з шприц-ручки Ліксумія®.
На рисунках, наведених нижче, показано, як змінюється активаційне віконце на кнопці для ін'єкцій шприц-ручки після активації.
|
Нова шприц-ручка (віконце має помаранчевий колір)
|
Шприц-ручка, готова для виконання ін'єкцій (віконце має білий колір)
|


Шприц-ручка активована і готова для виконання ін'єкцій. Після активації віконце залишається білим.
Як активувати нову шприц-ручку Ліксумія®
Крок 1. Зніміть ковпачок та перевірте шприц-ручку




Візуально оцініть рідину. Вона має бути прозорою та безбарвною і без сторонніх часточок. Якщо це не так, таку ручку використовувати не слід.
Якщо рідина не відповідає вищезгаданим вимогам, повідомте про це лікаря, фармацевта або медсестру.
Крок 2. Під'єднайте голку та зніміть з неї ковпачки

Для активації завжди слід використовувати нову голку.
Зніміть захисну мембрану з зовнішнього ковпачка голки.
Тримайте голку на одній лінії з шприц-ручкою. Тримайте її рівно під час загвинчування.
Збережіть
Викиньте
Слід обережно поводитися з відкритою голкою, щоб не поранити себе.
Зніміть з голки зовнішній та внутрішній ковпачки. Слід зберегти зовнішній ковпачок – він знадобиться пізніше для від'єднання голки.
Крок 3. Потягніть за кнопку для ін'єкцій
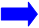

Сильно потягніть за кнопку для ін'єкцій до її зупинення


Після цього стрілочка буде вказувати в бік голки.
Крок 4. Натисніть та тримайте кнопку в цьому положенні, щоб видалити надлишок рідини
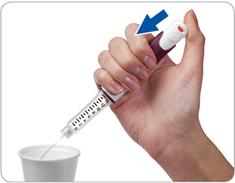
Спрямуйте голку в придатну ємність (наприклад, у паперову чашечку або згорнуту серветку) для того, щоб випустити рідину з тим, щоб потім цю ємність з надлишковою рідиною можна було викинути.
Натисніть на кнопку для ін'єкцій до кінця. Ви маєте відчути або почути характерне «клацання».
Не відпускайте кнопку для ін'єкцій 5 секунд, щоб дати стекти останнім краплям надлишкової рідини.
сек
Клац!
Якщо рідина не виходить з ручки, зверніться до розділу «Питання та відповіді».
Переконайтеся, що активаційне віконце тепер має білий колір.
Крок 5. Тепер Ваша шприц-ручка активована
Не намагайтеся активувати шприц-ручку повторно.
Виконувати заміну голки між активацією та виконанням першої ін'єкції не потрібно.
ЩОДЕННЕ ЗАСТОСУВАННЯ ШПРИЦ-РУЧКИ
Переходити до виконання процедур, описаних у цьому розділі, дозволяється лише за умови, що активаційне віконце вашої шприц-ручки має білий колір.
Слід застосовувати лише одну дозу цього лікарського засобу один раз на добу.

Крок А. Зніміть ковпачок та перевірте шприц-ручку

Візуально оцініть рідину. Вона має бути прозорою та безбарвною, без сторонніх часточок. Якщо це не так, таку ручку використовувати не слід.
У разі виявлення повітряних бульбашок зверніться до розділу «Питання та відповіді».
Перевірте кількість доз у шприц-ручці. Для цього подивіться на розташування чорного поршня відносно шкали, нанесеної на шприц-ручці.
Переконайтеся, що активаційне віконце має білий колір. Якщо колір активаційного віконця помаранчевий, перейдіть до розділу «Початок роботи» цієї інструкції.
Перевірте етикетку на шприц-ручці, щоб переконатися, що Ви застосовуєте саме потрібний лікарський засіб.
Крок B. Під'єднайте нову голку та зніміть з голки ковпачки.

Для кожної ін'єкції завжди слід використовувати нову голку.
Зніміть захисну мембрану з зовнішнього ковпачка голки.
Тримайте голку на одній лінії з шприц-ручкою. Тримайте її рівно під час загвинчування.
Викиньте
Збережіть

Слід обережно поводитися з відкритою голкою, щоб не поранити себе.
Зніміть з голки зовнішній та внутрішній ковпачки. Слід зберегти зовнішній ковпачок – він знадобиться пізніше для від'єднання голки.
Крок C. Сильно потягніть за кнопку для ін'єкцій до її зупинення.

Після цього стрілочка буде вказувати в бік голки.

Крок D. Для того щоб ввести дозу, натисніть та утримуйте в такому положенні кнопку для ін'єкцій.
Клац!
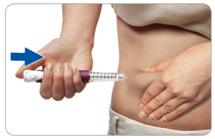
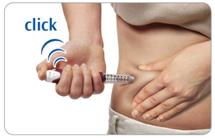
сек
Захопіть складку шкіри та введіть голку (в одну з ділянок шкіри, перелічених у розділі «Ділянки тіла, придатні для виконання ін'єкцій»).
Натисніть на кнопку для ін'єкцій до кінця. Ви маєте відчути або почути характерне «клацання».
Не відпускайте кнопку для ін'єкцій протягом 5 секунд, щоб повністю ввести дозу.
Витягніть голку зі шкіри.
Крок E. Від'єднуйте та викидайте голку після кожної ін'єкції.

Покладіть зовнішній ковпачок для голки на рівну поверхню. Спрямуйте голку усередину зовнішнього ковпачка.
Надягніть зовнішній ковпачок на шприц-ручку.

Стисніть зовнішній ковпачок, щоб захопити голку, і з його допомогою викрутіть голку з шприц-ручки.

Надягніть ковпачок на шприц-ручку.
Щоб дізнатися, як утилізувати використані голки, проконсультуйтеся з фармацевтом.
Крок F. Кожного разу при виконанні ін'єкції повторюйте всі кроки з розділу «ЩОДЕННЕ ЗАСТОСУВАННЯ ШПРИЦ-РУЧКИ».
Через 14 днів після активації шприц-ручку слід викинути. Це потрібно зробити, навіть якщо в шприц-ручці залишилася деяка кількість лікарського засобу. Проконсультуйтеся з лікарем або фармацевтом щодо утилізації шприц-ручки.
Графік дат активації та утилізації
Запишіть в цю таблицю дату активації шприц-ручки та дату, коли її необхідно утилізувати (через 14 днів після активації).
|
Шприц-ручка |
Дата активації |
Дата утилізації |
|
1 |
||
|
2 |
Зберігання
Загальна інформація
Тримайте шприц-ручки Ліксумія® в безпечному місці, де їх не зможуть побачити або дістати діти.
Уникайте потрапляння пилу та бруду на шприц-ручки Ліксумія®.
Після кожного використання закривайте шприц-ручку ковпачком для її захисту від світла.
Не дозволяється застосовувати шприц-ручку Ліксумія® після закінчення терміну придатності,
зазначеного на етикетці шприц-ручки та на картонній коробці. Кінцевою датою терміну
придатності вважається останній день зазначеного місяця.
Перед активацією шприц-ручки:
Ще не розпечатані шприц-ручки Ліксумія® слід зберігати в холодильнику при температурі від 2 до 8°C.
Не дозволяється заморожувати шприц-ручки Ліксумія® та застосовувати їх, якщо вони були
заморожені.
Перед використанням слід дати шприц-ручці нагрітися до кімнатної температури.
Після активації шприц-ручки:
- Активовану шприц-ручку Ліксумія® слід зберігати при температурі до 30°C. Не дозволяється заморожувати шприц-ручку Ліксумія® після її активації.
- Не слід зберігати шприц-ручку Ліксумія® з під'єднаною голкою. Коли шприц-ручку зберігають з під'єднаною голкою, це може призвести до контамінації або до можливого потрапляння в шприц-ручку повітря, що може негативно впливати на точність введеної дози.
- Після активації шприц-ручку Ліксумія® дозволяється використовувати протягом до 14 днів. Через 14 днів використану шприц-ручку Ліксумія® слід викинути. Це потрібно зробити, навіть якщо в шприц-ручці залишилася деяка кількість лікарського засобу.
Утилізація
- Перед тим як утилізувати шприц-ручку Ліксумія®, надягніть на неї ковпачок.
- Утилізуйте шприц-ручку Ліксумія®.
Поводження з препаратом
- Слід обережно поводитися зі шприц-ручкою Ліксумія®.
- Зовнішній корпус шприц-ручки Ліксумія® можна протирати вологою тканиною.
- Не дозволяється мочити, мити шприц-ручку Ліксумія® або наносити рідину на неї (змащувати), оскільки це може призвести до її пошкодження.
- Не слід використовувати шприц-ручку Ліксумія® при підозрі, що вона була пошкоджена. Не намагайтеся полагодити шприц-ручку самостійно.
Ділянки тіла, придатні для виконання ін'єкцій
Передня частина тіла Задня частина тіла
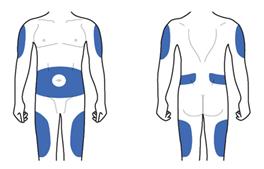
Шприц-ручка Ліксумія® призначена для виконання підшкірних ін'єкцій у будь-яку з ділянок тіла, позначених на рисунку вище синім кольором. Це стегна, живіт або верхня частина руки. Проконсультуйтеся з лікарем, як правильно робити ін'єкції.
Питання та відповіді
Що робити, якщо Ви забули активувати шприц-ручку Ліксумія® або зробили собі ін’єкцію до активації?
Якщо Ви зробили собі ін’єкцію перед активацією шприц-ручки, не намагайтеся виправити помилку, роблячи собі другу ін’єкцію. Зверніться до свого лікаря, фармацевта або медичної сестри, щоб отримати рекомендації з перевірки Вашого рівня цукру в крові.
Що робити, якщо у контейнері присутні бульбашки повітря?
Невеличкі бульбашки повітря у контейнері є нормальним явищем – вони не зашкодять Вам. Ваша доза буде вірною, і Ви можете дотримуватися інструкцій. Зверніться до свого лікаря, фармацевта або медичної сестри, якщо Вам потрібна буде допомога.
Що робити, якщо при активації з голки не виходить рідина?
Голка може бути забитою або неправильно приєднаною до шприц-ручки. Від’єднайте голку від шприц-ручки, приєднайте нову голку та повторіть лише Кроки 4 і 5. Якщо і після цього з голки не виходить рідина, Ваша шприц-ручка Ліксумія® може бути пошкоджена. Не використовуйте цей комплект Ліксумія®. Зверніться за допомогою до свого лікаря, фармацевта або медичної сестри.
Що робити, якщо ін’єкційна кнопка важко натискається протягом усієї ін’єкції?
Голка може бути забитою або неправильно приєднаною до шприц-ручки. Витягніть голку зі шкіри та від’єднайте голку від шприц-ручки. Приєднайте нову голку та повторіть лише Кроки D і E. Якщо і після цього з голки не виходить рідина, Ваша шприц-ручка Ліксумія® може бути пошкоджена. Не використовуйте цей комплект Ліксумія®. Зверніться за допомогою до свого лікаря, фармацевта або медичної сестри.
Побічні реакції.
Більш ніж 2600 пацієнтів одержували Ліксумія® окремо або в комбінації з метформіном, похідними сульфонілсечовини (комбіновано з метформіном або окремо), або з базальним інсуліном (окремо або комбіновано з метформіном, або інсуліну окремо або в комбінації з похідними сульфонілсечовини) в ході 8 масштабних клінічних досліджень з плацебо або з активним контролем фази ІІІ.
Частота побічних реакцій визначалися як: (дуже часто – ≥1/10; часто – ≥1/100 до <1/10; нечасто – ≥1/1000 до <1/100; рідко: ≥1/10000 до <1/1000; дуже рідко: <1/10000).
У кожній категорії «Система-Орган-Клас» побічні реакції представлені в порядку зниження їхньої частоти.
Таблиця 1. Побічні реакції, про які повідомлялося в ході плацебо- та активно контрольованих клінічних досліджень ІІІ фази протягом всього періоду терапії (включаючи період за межами основного 24-тижневого періоду терапії у дослідженнях з тривалістю ≥76 тижнів загальної терапії). В таблиці представлено побічні реакції, класифіковані по преференційному терміну, що відмічалися з частотою більше 5 %, якщо частота в групі Ліксумія® була вище, ніж в групах пацієнтів, що одержували препарати порівняння. Крім того, таблиця включає побічні реакції з частотою ≥ 2 % в групі Ліксумія®, якщо частота реакцій вдвічі перевищувала частоту реакцій в групі препаратів порівняння.
|
Система-Орган-Клас |
Частота виникнення |
||
|
Дуже часто |
Часто |
Нечасто |
|
|
Інфекційні та паразитарні захворювання |
Грип, інфекція верхніх дихальних шляхів, цистит, вірусна інфекція |
||
|
З боку імунної системи |
Анафілактична реакція |
||
|
Метаболічні та аліментарні розлади |
Гіпоглікемія (у комбінації з похідним сульфонілсечовини і/або базальним інсуліном) |
Гіпоглікемія (у комбінації лише з метформіном) |
|
|
З боку нервової системи |
Головний біль |
Запаморочення, сонливість |
|
|
Розлади з боку шлунково-кишкового тракту |
Нудота, блювання, діарея |
Диспепсія |
|
|
З боку шкіри та підшкірної клітковини |
Кропив'янка |
||
|
Система-Орган-Клас |
Частота виникнення |
||
|
Дуже часто |
Часто |
Нечасто |
|
|
З боку опорно-рухового апарату та сполучної тканини |
Біль у спині |
||
|
Загальні розлади та реакції у місці введення |
Свербіння у місці ін’єкції |
||
Опис окремих побічних реакцій.
Гіпоглікемія. У пацієнтів, які отримували препарат Ліксумія® як монотерапію, симптоматична гіпоглікемія спостерігалася у 1,7% пацієнтів, які отримували лікування ліксисенатидом, і у 1,6 % пацієнтів, що отримували плацебо. У випадку застосування препарату Ліксумія® у комбінації лише з метформіном симптомна гіпоглікемія за весь період лікування виникала у 7,0 % пацієнтів, які застосовували ліксисенатид, і у 4,8 % пацієнтів, які отримували плацебо.
У пацієнтів, які отримували препарат Ліксумія® в комбінації з сульфонілсечовиною та метформіном симптоматична гіпоглікемія за весь період лікування спостерігалася у 22,0 % пацієнтів, які отримували лікування ліксисенатидом, і у 18,4 % пацієнтів, які отримували плацебо (абсолютна різниця становила 3,6 %). У випадку застосування препарату Ліксумія® у комбінації з базальним інсуліном разом з метформіном або без нього симптомна гіпоглікемія за весь період лікування виникала у 42,1 % пацієнтів, які застосовували ліксисенатид, і у 38,9 % пацієнтів, які отримували плацебо (абсолютна різниця становила 3,2 %).
Протягом усього періоду терапії, коли препарат застосовували тільки з похідними сульфонілсечовини, симптоматична гіпоглікемія відмічалася у 22,7 % пацієнтів, які отримували лікування ліксисенатидом, порівняно з 15,2 % пацієнтів, які отримували плацебо (абсолютна різниця становила 7,5 %). Симптоматична гіпоглікемія відмічалася у 47,2 % пацієнтів, які отримували лікування ліксисенатидом у комбінації з похідними сульфонілсечовини та базальним інсуліном, в порівнянні з 21,6 % у пацієнтів групи плацебо (абсолютна різниця становила 25,6 %).
Загалом, частота виникнення тяжких симптоматичних гіпоглікемій була невисокою (0,4 % у пацієнтів групи препарату Ліксумія® та 0,2 % у пацієнтів групи плацебо) протягом усього періоду терапії в ході плацебо- та активно контрольованих клінічних досліджень ІІІ фази.
Розлади з боку шлунково-кишкової системи. Нудота та блювання були дуже часто згадуваними побічними реакціями протягом основного 24-тижневого періоду терапії. Частота виникнення нудоти була вищою в групі препарату Ліксумія® (26,1 %) у порівнянні з групою плацебо (6,2 %), частота блювання була вищою в групі препарату Ліксумія® (10,5 %) у порівнянні з групою плацебо (1,8 %). Ці реакції відзначалися здебільшого легким та скороминучим характером та виникали протягом перших 3 тижнів після початку терапії. В подальшому частота прогресивно знижувалася протягом наступних тижнів.
Реакції в місці введення. Протягом основного 24-тижневого періоду терапії реакції в місці введення спостерігалися у 3,9 % пацієнтів, що одержували препарат Ліксумія®, в той час як у пацієнтів групи плацебо такі реакції спостерігалися у 1,4 %. Більшість реакцій були легкої інтенсивності та зазвичай не призводили до відміни препарату.
Імуногенність. Відповідно до потенційних імуногенних властивостей лікарських засобів, що містять білки або пептиди, внаслідок терапії препаратом Ліксумія® у пацієнтів можуть вироблятися антитіла до ліксисенатиду.
Однак зміна рівня HbA1c щодо вихідного рівня була співставною незалежно від реакції на антитіла (позитивної чи негативної).
Статус антитіл (позитивний або негативний) в окремого пацієнта не дає можливості передбачити ступінь зниження HbA1c.
Відмінності загального профілю безпеки були відсутні у пацієнтів незалежно від реакції на антитіла, за винятком підвищення частоти виникнення реакцій в місці введення у пацієнтів з позитивною реакцією на антитіла (4,7% пацієнтів з позитивним результатом аналізу на антитіла порівняно з 2,5% пацієнтів з негативним результатом цього аналізу протягом усього періоду лікування). Більшість реакцій в місці введення були легкими незалежно від реакції на антитіла.
Алергічні реакції. Алергічні реакції, можливо пов’язані з ліксисенатидом, (такі як анафілактичні реакції, ангіоневротичні набряки, кропив’янка) спостерігалися у 0,4 % пацієнтів в групі ліксисенатиду, порівняно з менш ніж 0,1% пацієнтів, які отримували плацебо протягом основного 24-тижневого періоду терапії. Анафілактичні реакції відмічалися у 0,2 % пацієнтів, які отримували лікування ліксисенатидом, порівняно з жодним пацієнтом у групі плацебо. Більшість цих зареєстрованих алергічних реакцій були легкого ступеня тяжкості.
Протягом клінічних досліджень з вивчення ліксисенатиду був зареєстрований один випадок анафілактоїдної реакції.
Відміна. Протягом основного 24-тижневого періоду терапії частота відміни терапії у зв’язку з побічними явищами становила 7,4 % в групі препарату Ліксумія® в порівнянні з 3,2 % в групі плацебо.
Передозування. При застосуванні 30 мкг ліксисенатиду двічі на день протягом 13 тижнів дослідження спостерігалося лише підвищення частоти виникнення розладів з боку шлунково-кишкової системи.
При передозуванні слід розпочати відповідну підтримуючу терапію згідно з клінічними симптомами у пацієнта, а доза препарату Ліксумія® має бути знижена до призначеної дози.
Застосування у період вагітності або годування груддю.
Жінки репродуктивного віку. Препарат Ліксумія® не рекомендований до застосування у жінок репродуктивного віку, які не використовують контрацептивні засоби.
Вагітність. Доказові дані щодо застосування препарату у вагітних жінок відсутні. Дослідження на тваринах виявили ознаки токсичного впливу на репродуктивну систему. Потенційний ризик для людини невідомий. Препарат Ліксумія® не слід призначати у період вагітності; замість цього рекомендується застосування інсуліну. Якщо пацієнтка бажає завагітніти або вагітність вже настала застосування препарату треба припинити.
Період годування груддю. Невідомо, чи виводиться Ліксумія® з грудним молоком. З огляду на відсутність досвіду застосування препарату, його не слід призначати у період годування груддю.
Діти. Безпека та ефективність препарату Ліксумія® не досліджувалися у пацієнтів до 18 років.
Особливості застосування. Оскільки досвід застосування препарату ліксисенатиду у пацієнтів із цукровим діабетом 1 типу відсутній, його не слід призначати пацієнтам цієї групи. Препарат не слід застосовувати для лікування діабетичного кетоацидозу.
Гострий панкреатит. Застосування агоністів рецепторів глюкагоноподібного пептиду-1 (ГПП-1) був пов’язаний з ризиком розвитку гострого панкреатиту. Пацієнтів слід проінформувати про характерні симптоми панкреатиту: постійний сильний біль у животі. У разі підозри на панкреатит ліксисенатид відміняють. У разі підтвердження гострого панкреатиту застосування ліксисенатиду не відновлюють. У пацієнтів з панкреатитом в анамнезі препарат застосовують з обережністю. У пацієнтів з панкреатитом в анамнезі слід дотримуватися обережності при призначенні препарату.
Тяжкі шлунково-кишкові захворювання. Застосування агоністів рецепторів ГПП-1 може бути асоційоване з побічними реакціями з боку шлунково-кишкової системи. Застосування ліксисенатиду не досліджувалося у пацієнтів з тяжкими шлунково-кишковими розладами, включаючи тяжкий гастропарез, отже, застосування препарату у таких пацієнтів не рекомендоване.
Ниркова недостатність. Терапевтичний досвід застосування препарату у пацієнтів з помірною ниркової недостатністю (кліренс креатиніну: 30–50 мл/хв) обмежений, а терапевтичний досвід його використання у пацієнтів з важкою нирковою недостатністю (кліренс креатиніну менше 30 мл/хв) або на термінальних стадіях захворювань нирок відсутній. Ліксумія® має використовуватися з обережністю у пацієнтів з помірною нирковою недостатністю. Препарат не рекомендується використовувати у пацієнтів з важкою нирковою недостатністю та на термінальних стадіях захворювань нирок (див. розділи «Взаємодія з іншими лікарськими засобами та інші види взаємодій» та «Фармакокінетика»).
Гіпоглікемія. У пацієнтів, що застосовують препарат Ліксумія® у комбінації з похідними сульфонілсечовини або з базальним інсуліном, виникає підвищений ризик гіпоглікемії. Для його зменшення доцільно знизити дози похідних сульфонілсечовини або базального інсуліну (див. розділ «Спосіб застосування та дози»). Препарат Ліксумія® не можна призначати у комбінації з базальним інсуліном і похідними сульфонілсечовини через підвищення ризику виникнення гіпоглікемії.
Супутні лікарські засоби. Сповільнення спорожнення шлунку через використання ліксисенатиду може зменшувати швидкість абсорбції лікарських засобів, що приймаються перорально. Ліксумія має використовуватися з обережністю у пацієнтів, які приймають пероральні лікарські засоби, що потребують швидкої абсорбції у шлунково-кишковому тракті. Застосування таких препаратів одночасно з препаратом Ліксумія® потребує ретельного клінічного контролю або має вузьке терапевтичне вікно. Конкретні рекомендації стосовно прийому таких лікарських засобів надані у розділі «Взаємодія з іншими лікарськими засобами та інші види взаємодій».
Недосліджені популяції. Ліксисенатид не вивчався в комбінації з інгібіторами дипептидилпептидази 4 (ДПП-4). Досвід застосування препарату у пацієнтів із застійною серцевою недостатністю обмежений.
Дегідратація. Пацієнтів, які отримують препарат Ліксумія®, необхідно повідомити про потенційний ризик дегідратації, пов’язаної з побічними реакціями з боку шлунково-кишкового тракту, та вжити заходів для уникнення гіповолемії.
Допоміжні речовини. Цей лікарський засіб містить метакрезол, який може викликати алергічні реакції.
Здатність впливати на швидкість реакції при керуванні автотранспортом або роботі з іншими механізмами. Вплив препарату Ліксумія® на здатність керувати транспортними засобами або працювати з механізмами відсутній або незначний. При комбінованому застосуванні препарату Ліксумія® з похідними сульфонілсечовини або базальним інсуліном пацієнти повинні бути попереджені про необхідність уникати гіпоглікемії при керуванні транспортними засобами та роботі з механізмами.
Взаємодія з іншими лікарськими засобами та інші види взаємодій. Ліксисенатид є пептидом та не метаболізується цитохромом Р450. В дослідженнях in vitro ліксисенатид не впливав на активність досліджуваних ізоферментів цитохрому Р450 та транспортних систем.
Затримка випорожнення шлунка, спричинена препаратом Ліксумія®, може впливати на абсорбцію лікарських засобів для перорального застосування. За пацієнтами, які отримують лікарські засоби, що мають вузьке терапевтичне вікно або вимагають ретельного клінічного моніторингу, слід уважно спостерігати, особливо на початку лікування ліксисенатидом. Ці лікарські засоби необхідно приймати стандартним чином стосовно прийому ліксисенатиду. Якщо такі лікарські засоби повинні прийматися разом з їжею, пацієнтам слід порекомендувати (за можливості) застосовувати їх під час того прийому їжі, коли ліксисенатид не призначається.
Пацієнтам слід рекомендувати застосовувати пероральні лікарські засоби, такі як антибіотики, ефективність яких особливо залежить від порогової концентрації, щонайменше за 1 годину до або через 4 години після введення ліксисенатиду. Парацетамол. При одноразовому застосуванні 1000 мг парацетамолу AUC та t1/2 парацетамолу залишалися незмінними незалежно від часу застосування препарату (до або після застосування ліксисенатиду).
З огляду на наведені дані необхідність у корекції доз парацетамолу відсутня, але подовження tmax, яке спостерігалося, коли парацетамол призначався через 1 або4 години (2,0 та 1,75 год. відповідно) після прийому ліксисенатиду, слід враховувати, коли для ефективності необхідний швидкий початок дії.
Пероральні контрацептивні засоби.
Прийом перорального контрацептиву (етинілестрадіол 0,03 мг/ левоноргестрелу 0,15 мг) через 1 або 4 години після застосування ліксисенатиду не впливав на AUC і t1/2 етинілестрадіолу та левоноргестрелу, хоча Cmax етинілестрадіолу знижувалася на 52 % і 39 % відповідно, і Cmaxлевоноргестрелу зменшувалась на 46 % і 20 % відповідно, а середнє значення tmax збільшувалася на 1-3 години.
Зниження Cmax характеризується невисокою клінічною значущістю, отже, необхідність у корекції дози пероральних контрацептивних засобів відсутня.
Аторвастатин. Необхідність у корекції дози аторвастатину при його супутньому застосуванні з ліксисенатидом відсутня.
Варфарин та інші похідні кумарину. Необхідність в корекції дози варфарину при його супутньому застосуванні з ліксисенатидом відсутня, однак на початку та по закінченню лікування ліксисенатидом рекомендується частий контроль МНС у пацієнтів, які приймають варфарин і/або похідні кумарину.
Дігоксин. Необхідність в корекції дози дігоксину при його одночасного застосуванні з ліксисенатидом відсутня.
Раміприл. Необхідність у корекції дози раміприлу при його одночасному застосуванні з ліксисенатидом відсутня.
Фармакологічні властивості.
Механізм дії. Ліксисенатид являє собою потужний та селективний агоніст рецепторів ГПП-1. Рецептори ГПП-1 є мішенню природного ГПП-1, ендогенного гормону інкретину, який потенціює глюкозозалежну секрецію інсуліну β-клітинами підшлункової залози.
Дія ліксисенатиду опосередкована специфічною взаємодією препарату з рецепторами ГПП-1, що призводить до підвищення рівня внутрішньоклітинного циклічного аденозину монофосфату (цАМФ). Ліксисенатид стимулює секрецію інсуліну при підвищенні рівня глюкози крові, але не при нормальному рівні глюкози, що обмежує ризик гіпоглікемії. Паралельно пригнічується секреція глюкагону. При гіпоглікемії рятувальний механізм секреції глюкагону зберігається. Ліксисенатид гальмує випорожнення шлунка, таким чином знижуючи ступінь переносу глюкози, що міститься в їжі, в систему кровообігу. Фармакодинаміка. При застосуванні один раз на добу ліксисенатид покращує глікемічний контроль шляхом миттєвого та тривалого ефектів зниження концентрації глюкози як після їжі, так і натщесерце у пацієнтів із цукровим діабетом 2 типу.
Вплив препарату на постпрандіальний рівень глюкози був підтверджений в ході 4-тижневих клінічних досліджень в порівнянні з ліраглютидом в дозі 1,8 мг. Зниження від вихідного рівня показника AUC 0:30-4:30h глюкози плазми крові після пробного прийому їжі становило: ‑227,25 год·мг/дл (-12,61 год·ммоль/л) у групі лікування ліксисенатидом і -72,83 год·мг/дл (-4,04 год·ммоль/л) у групі лікування ліраглутидом.
Клінічна ефективність. Вплив препарату Ліксумія® на глікемічний контроль оцінювали в ході 6 рандомізованих, подвійних сліпих плацебо-контрольованих клінічних досліджень та одного рандомізованого, відкритого дослідження з активним контролем у порівнянні з ексенадитом. В цих дослідженнях брали участь 3825 пацієнтів (48,2 % - чоловіки та 51,8 % − жінки) з цукровим діабетом типу 2 (2445 пацієнтів було рандомізовано до групи ліксисенатиду).
768 пацієнтів (з яких 447 було рандомізовано до групи ліксисенатиду) були у віці від 65 років та старше, а 103 пацієнти (з яких 57 було рандомізовано до групи ліксисенатиду) були у віці від 75 років.
У ході завершених клінічних досліджень ІІІ фази було продемонстровано, що більш ніж 90 % пацієнтів були здатні залишатися на підтримуючій дозі препарату Ліксумія® у 20 мкг один раз на добу наприкінці основного 24-тижневого періоду терапії.
Додаткова терапія пероральними протидіабетичними засобами. Для застосування препарату Ліксумія® одночасно з метформіном, похідними сульфонілсечовини, піоглітазоном або з комбінацією цих лікарських засобів показано статистично значуще зниження рівня HbA1c, рівня глюкози натщесерце та 2-годинного постпрандіального рівня глюкози після тестового прийому їжі в порівнянні з плацебо наприкінці основного 24-тижневого періоду терапії (Таблиці 2 та 3). Зниження HbA1c було статистично значущим при застосуванні препарату один раз на добу незалежно від того, застосовувався препарат вранці або ввечері.
Цей вплив на показник HbA1c зберігався у довгострокових дослідженнях тривалістю до 76 тижнів.
Як додаткова терапія до метформіну окремо.
Таблиця 2. Плацебо-контрольовані клінічні дослідження в комбінації з метформіном (результати після 24 тижнів терапії).
|
Метформін як основна терапія |
|||||
|
Ліксисенатид 20 мкг |
Плацебо (N=159) |
Ліксисенатид 20 мкг |
Плацебо (N=170) |
||
|
Двоетапне початкове дозування* (N=160) |
Вранці (N=255) |
Ввечері (N=255) |
|||
|
Середній рівеньHbA1c(%): Вихідний рівень Середня зміна (за методом найменших квадратів) відносно вихідного рівня |
7,99 -0,92 |
8,03 -0,42 |
8,07 -0,87 |
8,07 -0,75 |
8,02 -0,38 |
|
Пацієнти (%), що досягли рівня HbA1c < 7,0% |
47,4 |
24,1 |
43,0 |
40,6 |
22,0 |
|
Середня маса тіла (кг): Вихідний рівень Середня зміна (за методом найменших квадратів) відносно вихідного рівня |
90,30 -2,63 |
87,86 -1,63 |
90,14 -2,01 |
89,01 -2,02 |
90,40 -1,64 |
У дослідженні з активним контролем в кінці основного 24-тижневого періоду лікування спостерігалися зниження показника HbA1c на -0,79 % при застосуванні препарату Ліксумія® один раз на добу порівняно з ‑0,96 % на фоні застосування ексенатиду двічі на добу з середнім значенням різниці між групами лікування 0,17 % (95 % ДІ: 0,033, 0,297) і подібний відсоток пацієнтів, які досягли показника HbA1c нижче 7 %, у групі лікування ліксисенатидом (48,5 %) і групі лікування ексенатидом (49,8 %).
Частота виникнення нудоти становила 24,5 % у групі лікування ліксисенатидом порівняно з 35,1 % у групі лікування ексенатидом двічі на добу, а частота виникнення симптомної гіпоглікемії при лікуванні ліксисенатидом становила 2,5 % протягом основного 24-тижневого періоду лікування порівняно з 7,9 % у групі лікування ексенатидом.
Як додаткова терапія до похідних сульфонілсечовини окремо або в комбінації з метформіном.
Таблиця 3. Плацебо-контрольовані дослідження ліксисенатиду в комбінації з похідними сульфонілсечовини (результати після 24 тижнів терапії).
|
Похідні сульфонілсечовини як основна терапія з метформіном та без нього |
||
|
Ліксисенатид 20 мкг (N=570) |
Плацебо (N=286) |
|
|
Середній рівень HbA1c (%): Вихідний рівень Середня зміна (за методом найменших квадратів) відносно вихідного рівня |
8,28 -0,85 |
8,22 -0,10 |
|
Пацієнти (%), що досягли рівня HbA1c < 7,0 % |
36,4 |
13,5 |
|
Середня маса тіла (кг): Вихідний рівень Середня зміна (за методом найменших квадратів) відносно вихідного рівня |
82,58 -1,76 |
84,52 -0,93 |
Як додаткова терапія до піоглітазону окремо або в комбінації з метформіном. У клінічному дослідженні додавання ліксисенатиду до піоглітазону у комбінації з метформіном або без нього у пацієнтів з недостатнім контролем захворювання при прийомі піоглітазону в кінці основного 24-тижневого періоду лікування призводило до зниження HbA1c від вихідного рівня на 0,90 % порівняно зі зниженням цього показника від вихідного рівня у групі плацебо на 0,34 %. У кінці основного 24-тижневого періоду лікування 52,3 % пацієнтів, які отримували ліксисенатид, досягли показника HbA1c нижче 7% порівняно з 26,4 % у групі плацебо.
Протягом основного 24-тижневого періоду лікування нудота відмічалася у 23,5 % пацієнтів у групі лікування ліксисенатидом порівняно з 10,6 % пацієнтів у групі плацебо, а симптомна гіпоглікемія спостерігалася у 3,4 % пацієнтів, які отримували ліксисенатид, порівняно з 1,2 % пацієнтів у групі плацебо.
Як додаткова терапія до базального інсуліну.Застосування препарату Ліксумія® додатково до базального інсуліну окремо або в комбінації з базальним інсуліном та метформіном, або в комбінації з базальним інсуліном та похідними сульфонілсечовини призводить до клінічно та статистично значущого зниження рівнів НвА1с та 2-годинного постпрандіального рівня глюкози після тестового прийму їжі в порівнянні з плацебо.
Таблиця 4. Плацебо-контрольовані дослідження в комбінації з базальним інсуліном (результати після 24 тижнів терапії).
|
Базальний інсулін як основна терапія |
Базальний інсулін як основна терапія |
|||
|
Окремо або в комбінації з метформіном |
Окремо або в комбінації з похідними сульфонілсечовини* |
|||
|
Ліксисенатид 20 мкг (N=327) |
Плацебо (N=166) |
Ліксисенатид 20 мкг (N=154) |
Плацебо (N=157) |
|
|
Середній рівень HbA1c (%): Вихідний рівень Середня зміна (за методом найменших квадратів) відносно вихідного рівня |
8,39 -0,74 |
8,38 -0,38 |
8,53 -0,77 |
8,53 0,11 |
|
Пацієнти (%) з досягнутим рівнем HbA1c < 7,0% |
28,3 |
12,0 |
35,6 |
5,2 |
|
Середня тривалість лікування базальним інсуліном на початок дослідження (роки) |
3,06 |
3,2 |
2,94 |
3,01 |
|
Середня зміна дози базального інсуліну (В ОД): Вихідний рівень Середня зміна (за методом найменших квадратів) відносно вихідного рівня |
53,62 -5,62 |
57,65 -1,93 |
24,87 -1,39 |
24,11 -0,11 |
|
Середня маса тіла (кг): Вихідний рівень Середня зміна (за методом найменших квадратів) відносно вихідного рівня |
87,39 -1,80 |
89,11 -0,52 |
65,99 -0,38 |
65,60 0,06 |
*Проведено на азіатській популяції.
Клінічне дослідження проводилося за участю пацієнтів, які раніше не отримували інсулін, з недостатнім контролем захворювання при застосуванні пероральних протидіабетичних засобів. Це дослідження складалося з 12-тижневого ввідного періоду з початком введення і титруванням дози інсуліну гларгін і 24-тижневого періоду лікування, протягом якого пацієнти отримували ліксисенатид або плацебо у комбінації з інсуліном гларгін і метформіном разом з тіазолідиндіоном або без нього. Протягом цього періоду постійно проводилося титрування дози інсуліну гларгін.
Протягом 12-тижневого ввідного періоду додаткове призначення і титрування дози інсуліну гларгін призводило до зниження показника HbA1cприблизно на 1 %. Додавання ліксисенатиду спричинило статистично значуще додаткове зниження HbA1c на 0,71 % у групі лікування ліксисенатидом порівняно з 0,40 % у групі плацебо. У кінці 24-тижневого періоду лікування 56,3 % пацієнтів, які отримували ліксисенатид, досягли показника HbA1c нижче 7 % порівняно з 38,5 % пацієнтів у групі плацебо.
Протягом 24‑тижневого періоду лікування 22,4% пацієнтів, які отримували ліксисенатид, відмітили щонайменше один епізод симптоматичної гіпоглікемії порівняно з 13,5 % пацієнтів у групі плацебо. Частота виникнення гіпоглікемії переважно збільшувалася у групі лікування ліксисенатидом протягом перших 6 тижнів лікування, а в подальшому була такою ж, як у групі плацебо.
Рівень глюкози натщесерце. Наприкінці основного 24-тижневого періоду терапії в ході плацебо-контрольованих клінічних досліджень зниження рівнів глюкози натщесерце, досягнуте терапією препаратом Ліксумія®, склало від 0,42 ммоль/л до 1,19 ммоль/л у порівнянні з вихідним рівнем.
Постпрандіальний рівень глюкози. Терапія препаратом Ліксумія® призвела до статистично значущого зниження 2-годинного постпрандіального рівня глюкози після тестового прийому їжі в порівнянні з плацебо незалежно від типу основної терапії.
Наприкінці основного 24-тижневого періоду терапії зниження рівня глюкози, викликане препаратом Ліксумія®, коливалося від 4,51 ммоль/л до 7,96 ммоль/л відносно вихідного рівня в рамках всіх досліджень, в ході яких проводили визначення постпрандіального рівня глюкози; у 26,2-46,8 % пацієнтів зміна 2-годинного постпрандіального рівня глюкози була нижче 7,8 ммоль/л.
Маса тіла. Застосування препарату Ліксумія® в комбінації з метформіном та/або з сульфонілсечовиною призводило до стійкої зміни маси тіла від вихідного рівня в усіх контрольованих дослідженнях в межах від -1,76 кг до -2,96 кг в кінці основного 24-тижневого періоду лікування. Зміна маси тіла від вихідного рівня в межах від -0,38 кг до -1,80 кг також спостерігалася у пацієнтів, які отримували ліксисенатид і стабільну дозу базального інсуліну у якості монотерапії або у комбінації з метформіном або похідним сульфонілсечовини. У пацієнтів, яким вперше призначили інсулін, маса тіла майже не змінювалася в групі лікування ліксисенатидом, тоді як в групі плацебо спостерігалося її збільшення. Зниження маси тіла зберігалося в довгострокових дослідженнях тривалістю до 76 тижнів.
Зниження маси тіла не залежало від частоти виникнення нудоти та блювання.
Функція β-клітин. У клінічних дослідженнях препарат Ліксумія® покращував функцію β-клітин, згідно з оцінкою методом моделі оцінки гомеостазу для функції β-клітин (індекс HOMA).
Відновлення першої фази секреції інсуліну та покращення другої фази секреції інсуліну у відповідь на внутрішньовенне болюсне введення глюкози було продемонстровано у пацієнтів з цукровим діабетом типу 2 (n=20) після одноразового введення препарату Ліксумія®.
Оцінка серцево-судинних функцій. У жодних плацебо-контрольованих дослідженнях III фази у пацієнтів з цукровим діабетом 2 типу не спостерігалося підвищення середнього значення частоти серцевих скорочень.
У плацебо-контрольованих дослідженнях III фази відмічалося зниження середнього систолічного і діастолічного артеріального тису на величини до 2,1 мм рт. ст. і 1,5 мм рт. ст. відповідно.
Метааналіз усіх оцінюваних незалежними експертами серцево-судинних подій (смерть від серцево-судинної патології, нефатальний інфаркт міокарда, нефатальний інсульт, госпіталізація з приводу нестабільної стенокардії, госпіталізація з приводу серцевої недостатності і процедура реваскуляризації коронарних артерій), які спостерігалися в 8 плацебо-контрольованих дослідженнях III фази, що включили 2673 пацієнти з цукровим діабетом 2 типу, які отримували лікування ліксисенатидом, і 1448 пацієнтів, які отримували плацебо, показав відношення ризиків 1,03 (95 % довірчий інтервал 0,64; 1,66) для ліксисенатиду порівняно з плацебо. Кількість цих подій у клінічних дослідженнях була низькою (1,9 % у пацієнтів, які отримували лікування ліксисенатидом, і 1,8 % у пацієнтів з групи плацебо), що не дає можливості зробити які-небудь чіткі висновки. Частота виникнення окремих СС подій (ліксисенатид порівняно з плацебо) була такою: смерть від серцево-судинної патології (0,3 % порівняно з 0,3 %), нефатальний інфаркт міокарда (0,4 % порівняно з 0,4 %), нефатальний інсульт (0,7 % порівняно з 0,4 %), госпіталізація з приводу нестабільної стенокардії (нуль порівняно з 0,1 %),госпіталізація з приводу серцевої недостатності (0,1 % порівняно з нулем) і процедура реваскуляризації коронарних артерій (0,7 % порівняно з 1,0 %).
Діти. Європейське агентство лікарських засобів відклало зобов’язання надати результати досліджень з вивчення препарату Ліксумія® у одній або більше субпопуляцій дітей з цукровим діабетом 2 типу (інформація щодо застосування препарату у дітей наведена у розділі «Діти»).
Фармакокінетика.
Абсорбція.Після підшкірного введення препарату пацієнтам з цукровим діабетом типу 2 абсорбція ліксисенатиду протікає швидко і не залежить від застосованої дози. Незалежно від дози та від того, чи введений ліксисенатид одноразово чи в декількох дозах, медіанний tmax у пацієнтів з цукровим діабетом становить від 1 до 3,5 години. Відсутні клінічно значущі відмінності в швидкості абсорбції ліксисенатиду після підшкірного введення у ділянку живота, стегна або руки.
Розподіл.Ліксисенатид характеризується середнім (55 %) рівнем зв’язування з білками плазми людини. Уявний об’єм розподілу після підшкірного введення ліксісенатиду (Vz/F) становить приблизно 100 л.
Біотрансформація і виведення.Як всі білки, ліксисенатид виводиться шляхом клубочкової фільтрації з подальшою канальцевою реабсорбцією та подальшою метаболічною деградацією до пептидів меншої маси та амінокислот, які знову приєднуються до білкового метаболізму.
Після багаторазового введення у пацієнтів з цукровим діабетом 2 типу середній термінальний час напіввиведення становив близько 3 годин, а середній уявний кліренс (CL/F) був приблизно 35 л/год.
Особливі популяції.
Пацієнти з нирковою недостатністю. Відсутні суттєві відмінності середнього кліренсу, Cmax та AUC ліксисенатиду у пацієнтів з нормальною нирковою функцією та пацієнтів з нирковою недостатністю легкого та середнього ступеня тяжкості (кліренс креатиніну, розрахований за формулою Кокрофта-Голта, 50–80 мл/хв). У пацієнтів з помірною нирковою недостатністю (кліренс креатиніну 30–50 мл/хв) AUC збільшувалася на 24 %, а у пацієнтів з тяжкою нирковою недостатністю (кліренс креатиніну 15–30 мл/хв) – на 46 %.
Пацієнти з печінковою недостатністю. Оскільки ліксисенатид виводиться головним чином нирками, не проводилося жодних фармакокінетичних досліджень у пацієнтів з гострою або хронічною печінковою недостатністю. Не очікується, що порушення печінкової функції впливатимуть на фармакокінетику ліксисенатиду.
Стать. Стать не мала клінічно значущого впливу на фармакокінетику ліксисенатиду.
Раса. Етнічне походження не спричиняє клінічно значущого впливу на фармакокінетику ліксисенатиду, про що свідчать результати фармакокінетичних досліджень, проведених з участю пацієнтів європейського, японського та китайського походження.
Пацієнти літнього віку. Вік не спричиняє клінічно значущого впливу на фармакокінетику ліксисенатиду. У дослідженні фармакокінетики у людей похилого віку без цукрового діабету введення ліксисенатиду в дозі 20 мкг призводило до середнього підвищення AUC ліксисенатиду у цих осіб на 29 % (11 учасників віком 65–74 років і 7 учасників віком ≥75 років) порівняно з 18 учасниками віком від 18 до 45 років, що, ймовірно, пов’язане зі зниженням функції нирок у групі пацієнтів похилого віку.
Маса тіла. Маса тіла не мала клінічно значущого впливу на AUC ліксисенатиду.
Доклінічні дані з безпечності. За даними доклінічних досліджень з оцінки фармакологічних та токсикологічних властивостей препарату не виявлено жодних свідоцтв особливих ризиків для людини.
У дворічних дослідженнях канцерогенності при підшкірному введенні у мишей та щурів спостерігалися нелетальні С-клітинні пухлини щитоподібної залози, які вважалися зумовленими негенотоксичним механізмом, опосередкованим рецептором GLP‑1, до якого гризуни особливо чутливі. C-клітинна гіперплазія і аденома спостерігалися у щурів на фоні застосування усіх досліджуваних доз, а максимальну дозу відсутності спостережуваних побічних явищ (NOAEL) не можна було визначити. У мишей ці ефекти виникали при рівні експозиції, що у 9,3 рази перевищував експозицію у людей при застосуванні терапевтичних доз препарату. У мишей не спостерігалося С-клітинних карцином, а у щурів вони виникали при рівні експозиції, що приблизно у 900 разів перевищував експозицію у людей при застосуванні терапевтичних доз. У дворічному дослідженні канцерогенності при підшкірному введенні у мишей відмічено 3 випадки аденокарциноми в ендометрії зі статистично значущим підвищенням частоти цього захворювання в групі застосування середніх доз препарату, які створювали рівень експозиції, що перевищував експозицію у людей у 97 разів. Не було відмічено жодних ефектів, пов’язаних із застосуванням препарату.
Дослідження на тваринах не вказують на який-небудь безпосередній шкідливий вплив препарату на фертильність самців і самок щурів.
У собак, які отримували ліксисенатид, спостерігалися зворотні порушення у яєчках і придатках яєчників. У здорових чоловіків не відмічено жодного впливу препарату на сперматогенез, пов’язаний із застосуванням препарату.
У дослідженнях з вивчення впливу препарату на внутрішньоутробний розвиток у щурів на фоні усіх досліджуваних доз ліксисенатиду (за рівня експозиції, що у 5 разів перевищував експозицію у людей) і у кролів при введенні високих доз препарату (за рівня експозиції, що у 32 рази перевищував експозицію у людей) спостерігалися вади розвитку, сповільнення росту плода, затримка осифікації і побічні ефекти з боку скелета. В обох видів дослідних тварин відмічалася невелика токсична дія на материнський організм, що проявлялася у вигляді зменшення споживання їжі та зниження маси тіла. Ріст новонароджених дитинчат був сповільнений серед самців щурів, які піддавалися дії високих доз ліксисенатиду протягом пізнього гестаційного періоду і лактації, з невеликим збільшенням смертності щурят.
Фармацевтичні характеристики.
Основні фізико-хімічні властивості: прозора безбарвна рідина.
Термін придатності. 2 роки.
Термін придатності після першого відкриття упаковки:14 діб.
Умови зберігання. Зберігати у недоступному для дітей місці. Зберігати у холодильнику при температурі від +2°С до +8°С подалі від морозильного відділення. Не заморожувати. Умови зберігання після першого відкриття упаковки:зберігати при температурі не вище 30°С. Не охолоджувати. Не заморожувати. Закривати шприц-ручку ковпачком для захисту від світла.
Упаковка.
№ 1: по 3 мл у картриджі, вмонтованому у шприц-ручку фіолетового кольору, по 1 шприц-ручці в картонній коробці.
Категорія відпуску. За рецептом.
Виробник. Санофі-Авентіс Дойчланд ГмбХ, Німеччина.
Місцезнаходження. Брюнінгштрассе 50 Н600, H500, H590 65926 Франкфурт-на-Майні, Німеччина.
Аналоги
Совпадает код ATХ + действующие вещества + форма випуска
| Международное название | Lixisenatide - Ликсисенатид |
| Код АТХ | A10BX10 |
| Форма выпуска | раствор |
- Ликсумия 10 мкг Санофі-Авентіс Дойчланд ГмбХ, Німеччина
 Сучасна стратегія лікування хворих з артеріальною гіпертензією з високим ризиком ускладнень
Сучасна стратегія лікування хворих з артеріальною гіпертензією з високим ризиком ускладнень
 Рекомендації Української асоціації кардіологів з профілактики та лікування артеріальної гіпертензії
Рекомендації Української асоціації кардіологів з профілактики та лікування артеріальної гіпертензії
 Рекомендовані схеми діагностики та лікування пацієнтів з некоронарогенними хворобами та вадами серця
Рекомендовані схеми діагностики та лікування пацієнтів з некоронарогенними хворобами та вадами серця
 Медикоментозная профилактика инсульта при артериальной гипертензии (укр)
Медикоментозная профилактика инсульта при артериальной гипертензии (укр)