
Залтрап инструкция, аналоги и состав
| Показания: | ЗАЛТРАП (ZALTRAP) у комбінації з хіміотерапією іринотеканом/ 5-фторурацилом/фоліновою кислотою (схема FOLFIRI) показаний дорослим пацієнтам з метастатичним колоректальним раком (МКРР), який виявився резистентним до схеми хіміотерапії на основі оксаліплатину або прогресував на фоні її застосування. |
| Форма випуска: | Концентрат для розчину для інфузій, 25 мг/мл по 4 мл або 8 мл у флаконі |
| Производитель, страна: | Санофі-Авентіс Дойчланд ГмбХ, Німеччина |
| Действующее вещества: | 1 флакон містить: афліберсепту 100 мг/4 мл або 200 мг/8 мл |
| МНН: | Aflibercept - Афлиберсепт |
| Регистрация: | UA/13061/01/01з 30.12.2013 по 30.12.2018. Приказ 789 від 27.11.2015 |
| Код АТХ: |
Загальна характеристика
Міжнародна непатентована назва: aflibercept.
Основні властивості лікарської форми:
Препарат ЗАЛТРАП (ZALTRAP) є стерильним, апірогенним розчином для внутрішньовенного введення з концентрацією афліберсепту 25 мг/мл без консервантів у флаконах з прозорого скла по 5 мл або 10 мл. Зовнішній вигляд препарату: прозорий розчин від безбарвного до блідо-жовтого кольору.
Якісний та кількісний склад
1 флакон містить:
Діюча речовина – афліберсепту 25 мг/ мл (афліберсепту 100 мг / 4 мл або 200 мг / 8 мл).
Допоміжні речовини – натрію фосфат одноосновний, моногідрат; натрію фосфат двоосновний, гептагідрат; лимонна кислота, моногідрат; натрію цитрат, дигідрат; натрію хлорид гранульований; хлористоводнева кислота 36%; натрію гідрооксид; сахароза; полісорбат 20; вода для ін’єкцій.
Форма випуску
Концентрат для розчину для інфузій, 25 мг / мл, по 4 мл або по 8 мл у флаконі.
Код ATC. L01XX. Інші протипухлинні засоби.
Імунологічні і біологічні властивості
Фармакодинамічні властивості.
Механізм дії. Фактори росту ендотелію судин A та B (Vascular Endothelial Growth Factors A, В; VEGF-A, VEGF-B) та плацентарний фактор росту (Placental Growth Factor; PlGF) належать до родини ангіогенних факторів VEGF, які можуть виявляти потужну мітогенну і хемотактичну дію та є факторами підвищення судинної проникності шляхом впливу на ендотеліальні клітини. Дія VEGF-A опосередкована двома рецепторними тирозинкіназами – VEGFR-1 та VEGFR-2, які розташовані на поверхні ендотеліальних клітин. PlGF та VEGF-B зв'язуються тільки з рецептором VEGFR-1, який також є на поверхні лейкоцитів. Надлишкова активація цих рецепторів фактором VEGF-A може призвести до патологічної неоваскуляризації та надлишкової проникності судин. Фактор PlGF також пов'язаний з патологічною неоваскуляризацією та рекрутингом клітин запалення до пухлин.
Афліберцепт, також відомий в науковій літературі під назвою VEGF TRAP («пастка для VEGF»), – це рекомбінантний химерний білок, який складається з VEGF-зв'язувальних фрагментів позаклітинних доменів рецепторів 1 та 2 VEGF людини та Fc-фрагменту імуноглобуліна IgG1 людини, поєднаних між собою. Афліберцепт отримують за допомогою системи експресування генів ссавців у культурі клітинної лінії K-1 яєчників китайського хом'ячка (CHO) на основі рекомбінантної ДНК. Афліберцепт є димерним глікопротеїном – його білкова частина має молекулярну масу 97 кілодальтон (кДа), а глікозильований залишок обумовлює додаткові 15% від загальної молекулярної маси, яка загалом становить 115 кДа.
Афліберсепт діє як розчинний рецептор-пастка, що зв'язується з VEGF-A з більшою афінністю, ніж його природні рецептори, а також зі спорідненими лігандами PlGF та VEGF-B. Діючи як пастка для лігандів, афліберцепт запобігає зв'язуванню ендогенних лігандів зі своїми когнатними рецепторами і таким чином блокує опосередковану цими рецепторами передачу сигналів.
Афліберсепт блокує активацію рецепторів VEGF та проліферацію ендотеліальних клітин і таким чином інгібує ріст нових судин, які постачають у пухлинні новоутворення кисень та поживні речовини.
Афліберсепт зв'язується з VEGF-A людини (рівноважна константа дисоціації KD = 0,5 пМ для VEGF-A165 та 0,36 пМ для VEGF-A121), PlGF людини (KD = 39 пМ для PlGF-2) та з VEGF-B людини (KD = 1,92 пМ) і утворює стійкий, інертний комплекс без виявленої біологічної активності.
Фармакодинамічні ефекти.
Афліберсепт, введений мишам з ксенотрансплантатами або алотрансплантатами пухлин, інгібував ріст різних видів злоякісних новоутворень.
Клінічна ефективність та безпечність. Ефективність та безпечність препарату ЗАЛТРАП (ZALTRAP) оцінювалися в ході рандомізованого, подвійного-сліпого, плацебо-контрольованого дослідження у пацієнтів з метастатичним колоректальним раком, які раніше отримували терапію на основі оксаліплатину, з попереднім застосуванням бевацизумабу або без нього. Всього в дослідженні взяли участь 1226 пацієнтів, які були рандомізовані (1:1) для отримання препарату ЗАЛТРАП (ZALTRAP) (N=612; 4 мг/кг у вигляді внутрішньовенної інфузії протягом 1 години в день 1) або плацебо (N=614) в комбінації з 5-фторурацилом та іринотеканом [схема FOLFIRI: одночасне введення у день 1 за допомогою системи для внутрішньовенних інфузій з Y-подібним конектором іринотекану 180 мг/м2 у вигляді внутрішньовенної інфузії протягом 90 хвилин та фолінової кислоти (рацемічна суміш право- та лівообертальних ізомерів) 400 мг/м2 у вигляді внутрішньовенної інфузії протягом 2 годин, з подальшим введенням 5-ФУ 400 мг/м2 у вигляді внутрішньовенного болюсу, а потім – 5-ФУ 2400 мг/м2 у вигляді безперервної внутрішньовенної інфузії протягом 46 годин]. Ці цикли хіміотерапії в обох групах повторювали кожні 2 тижні. Лікування продовжувалося до виявлення прогресування захворювання або до появи неприйнятних токсичних реакцій. Первинною кінцевою точкою при оцінюванні ефективності була загальна виживаність. Стратифікація пацієнтів проводилася за шкалою оцінки функціонального статусу ECOG (0, 1 чи 2 бали) та за критерієм попереднього застосування бевацизумабу (так чи ні).
Обидві групи були добре збалансовані за демографічними характеристиками (вік, расова приналежність, функціональний статус за шкалою ECOG та наявність чи відсутність застосування бевацизумабу в анамнезі). Медіана віку 1226 пацієнтів, рандомізованих для участі у дослідженні, становила 61 рік; 58,6% пацієнтів були чоловічої статі; функціональний статус за шкалою ECOG на вихідному рівні у 97,8% пацієнтів становив 0 або 1 бал і у 2,2% пацієнтів – 2 бали. З 1226 рандомізованих пацієнтів 89,4% та 90,2% пацієнтів, які приймали плацебо/FOLFIRI та ЗАЛТРАП (ZALTRAP)/FOLFIRI відповідно, раніше отримували комбіновану хіміотерапію на основі оксаліплатину для лікування метастатичних/поширених форм раку. Близько 10% пацієнтів (10,4% пацієнтів у групі плацебо/FOLFIRI та 9,8% пацієнтів у групі ЗАЛТРАП (ZALTRAP)/FOLFIRI) раніше отримували ад'ювантну хіміотерапію на основі оксаліплатину, і у них відбулося прогресування захворювання під час такої терапії або в межах 6 місяців після її закінчення. У 373 пацієнтів (30,4%) хіміотерапія на основі оксаліплатину застосовувалася в комбінації з бевацизумабом.
Результати оцінювання ефективності схем ЗАЛТРАП (ZALTRAP)/FOLFIRI та плацебо/FOLFIRI підсумовані на Рисунку 1 та в Таблиці 2.
Рисунок 1. Загальна виживаність (місяці) – Криві Каплана-Мейєра для досліджуваних груп – Популяція рандомізованих пацієнтів, які прийняли хоча б по одній дозі досліджуваного препарату (intention-to-treat, ITT)
|
|
||
|
Криві загальної виживаності Каплана-Мейєра (95% довірчий інтервал [ДІ]) (місяці) 25% квартиль Медіана 75% квартиль |
Плацебо/FOLFIRI 6,63 (6,24 – 7,59) 12,06 (11,07 –13,08) 21,03 (18,92 – 22,80) |
ЗАЛТРАП (ZALTRAP)/FOLFIRI 7,62 (6,60 – 8,46) 13,50 (12,52 – 14,95) 25,59 (22,01 – 31,70) |
|
Відношення ризиків (ВР) (95% ДІ) – 0,817 (0,714 - 0,935); p-значення за результатами лог-рангового тесту – 0,0032 |
||
|
Ймовірність виживання |
|
|
|
Час (місяці) |
||
 Плацебо/FOLFIRI
Плацебо/FOLFIRI ЗАЛТРАП (ZALTRAP)/FOLFIRI
ЗАЛТРАП (ZALTRAP)/FOLFIRI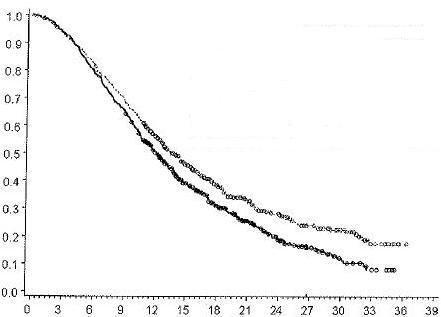
|
Кількість пацієнтів з ризиком смерті |
||||||||
|
Плацебо |
614 |
485 |
286 |
131 |
51 |
14 |
||
|
ЗАЛТРАП (ZALTRAP) |
612 |
498 |
311 |
148 |
75 |
33 |
||
|
Ймовірність виживання (%) |
||||||||
|
Плацебо |
79,1 |
50,3 |
30,9 |
18,7 |
12,0 |
|||
|
ЗАЛТРАП (ZALTRAP) |
81,9 |
56,1 |
38,5 |
28,0 |
22,3 |
|||
Таблиця 2. Основні кінцеві точки оцінки ефективностіa – Популяція ITT
|
Плацебо/FOLFIRI (N=614) |
ЗАЛТРАП (ZALTRAP)/FOLFIRI (N=612) |
|
|
Загальна виживаність (ЗВ) |
||
|
Кількість випадків смерті, n (%) |
460 (74,9%) |
403 (65,8%) |
|
Медіана загальної виживаності (95% ДІ) (місяці) |
12,06 (11,07 – 13,08) |
13,50 (12,52 – 14,95) |
|
Стратифіковане відношення ризиків (95% ДІ) |
0,817 (0,714 – 0,935) |
|
|
Стратифіковане p-значення за результатами лог-рангового тесту |
0,0032 |
|
|
Виживаність без прогресування захворювання (ВБП)b |
||
|
Кількість подій, n (%) |
454 (73,9%) |
393 (64,2%) |
|
Медіана ВБП (95% ДІ) (місяці) |
4,67 (4,21 – 5,36) |
6,90 (6,51 – 7,20) |
|
Стратифіковане відношення ризиків (95% ДІ) |
0,758 (0,661 – 0,869) |
|
|
Стратифіковане p-значення за результатами лог-рангового тесту |
0,00007 |
|
|
Загальна відповідь на терапію (повна відповідь + часткова відповідь) (95% ДІ) (%)c |
11,1 (8,5 – 13,8) |
19,8 (16,4 – 23,2) |
|
Стратифіковане p-значення за результатами використання критерію Кохрана-Мантеля-Гензеля |
0,0001 |
|
a Стратифікація за функціональним статусом за шкалою ECOG (0, 1 або 2 бали) та за прийомом бевацизумабу в анамнезі (так або ні).
b ВБП (за результатами оцінки пухлини Незалежним наглядовим комітетом): порогове значення для статистичної значущості встановлено на рівні 0,0001.
c Рівень загальної об'єктивної відповіді за результатами оцінки Незалежним наглядовим комітетом
Були виконані аналіз загальної виживаності (ЗВ) та виживаності без прогресування захворювання (ВБП) за стратифікаційними факторами. На фоні лікування комбінацією ЗАЛТРАП (ZALTRAP)/FOLFIRI у пацієнтів, які раніше отримували бевацизумаб, спостерігався кількісно менший лікувальний ефект щодо ЗВ у порівнянні із тими пацієнтами, які раніше не отримували бевацизумаб, без доказових даних на користь гетерогенності лікувального ефекту (результати тестування на наявність взаємодії виявилися статистично незначущими). Результати в залежності від попереднього застосування бевацизумабу підсумовані у Таблиці 3.
Таблиця 3. ЗВ та ВБП в залежності від попереднього застосування бевацизумабуа – Популяція ITT
|
Плацебо/FOLFIRI (N=614) |
ЗАЛТРАП (ZALTRAP)/FOLFIRI (N=612) |
|
|
Загальна виживаність |
||
|
Пацієнти з попереднім застосуванням бевацизумабу (n (%)) |
187 (30,5%) |
186 (30,4%) |
|
Медіана ЗВ (95% ДІ) (місяці) |
11,7 (9,96 – 13,77) |
12,5 (10,78 – 15,47) |
|
Відношення ризиків (95% ДІ) |
0,862 (0,676 – 1,100) |
|
|
Пацієнти без попереднього застосування бевацизумабу (n (%)) |
427 (69,5%) |
426 (69,6%) |
|
Медіана ЗВ (95% ДІ) (місяці) |
12,4 (11,17 – 13,54) |
13,9 (12,72 – 15,64) |
|
Відношення ризиків (95% ДІ) |
0,788 (0,671 – 0,925) |
|
|
Виживаність без прогресування захворювання |
||
|
Пацієнти з попереднім застосуванням бевацизумабу (n (%)) |
187 (30,5%) |
186 (30,4%) |
|
Медіана ВБП (95% ДІ) (місяці) |
3,9 (3,02 – 4,30) |
6,7 (5,75 – 8,21) |
|
Відношення ризиків (95% ДІ) |
0,661 (0,512 – 0,852) |
|
|
Пацієнти без попереднього застосування бевацизумабу (n (%)) |
427 (69,5%) |
426 (69,6%) |
|
Медіана ВБП (95% ДІ) (місяці) |
5,4 (4,53 – 5,68) |
6,9 (6,37 – 7,20) |
|
Відношення ризиків (95% ДІ) |
0,797 (0,679 – 0,936) |
|
a Визначалося за допомогою системи інтерактивної голосової відповіді
Також був виконаний аналіз ЗВ і ВБП за функціональним статусом за шкалою ECOG. Відношення ризиків (95% ДІ) для загальної виживаності становило 0,77 (0,64 – 0,93) в підгрупі пацієнтів з функціональним статусом на рівні 0 балів за шкалою ECOG і 0,87 (0,71 – 1,06) в підгрупі пацієнтів з функціональним статусом на рівні 1 бал за шкалою ECOG. Відношення ризиків (95% ДІ) для виживаності без прогресування захворювання становило 0,76 (0,63 – 0,91) в підгрупі пацієнтів з функціональним статусом на рівні 0 балів за шкалою ECOG і 0,75 (0,61 – 0,92) в підгрупі пацієнтів з функціональним статусом на рівні 1 бал за шкалою ECOG.
У Таблиці 4 підсумовані результати post hoc аналізів з виключенням пацієнтів, у яких відбулося прогресування захворювання на фоні отримання ад’ювантної терапії або протягом 6 місяців після її закінчення, для пацієнтів з попереднім застосуванням бевацизумабу або без нього.
Таблиця 4. Post hoc аналізи з виключенням пацієнтів, у яких відбулося прогресування захворювання на фоні отримання ад’ювантної терапіїa,b
|
Плацебо/FOLFIRI (N=550) |
ЗАЛТРАП (ZALTRAP)/FOLFIRI (N=552) |
|
|
Пацієнти з попереднім застосуванням бевацизумабу, з виключенням лише пацієнтів, у яких відбулося прогресування захворювання на фоні отримання ад’ювантної терапії (n (%)) |
179 (32,5%) |
177 (32,1%) |
|
Медіана загальної виживаності (95% ДІ) (місяці) |
11,7 (9,66 – 13,27) |
13,8 (11,01 – 15,87) |
|
Відношення ризиків (95% ДІ) |
0,812 (0,634 – 1,042) |
|
|
Медіана ВБП (95% ДІ) (місяці) |
3,9 (3,02 – 4,30) |
6,7 (5,72 – 8,21) |
|
Відношення ризиків (95% ДІ) |
0,645 (0,498 – 0,835) |
|
|
Пацієнти без попереднього застосування бевацизумабу, з виключенням лише пацієнтів, у яких відбулося прогресування захворювання на фоні отримання ад’ювантної терапії (n (%)) |
371 (67,5%) |
375 (67,9%) |
|
Медіана загальної виживаності (95% ДІ) (місяці) |
12,4 (11,17 – 13,54) |
13,7 (12,71 – 16,03) |
|
Відношення ризиків (95% ДІ) |
0,766 (0,645 – 0,908) |
|
|
Медіана ВБП (95% ДІ) (місяці) |
5,3 (4,50 – 5,55) |
6,9 (6,24 – 7,20) |
|
Відношення ризиків (95% ДІ) |
0,777 (0,655 – 0,921) |
|
a Визначалося за допомогою системи інтерактивної голосової відповіді
b Загальна виживаність в популяції ITT за виключенням пацієнтів, у яких відбулося прогресування захворювання на фоні ад’ювантної терапії або протягом 6 місяців після її закінчення, характеризувалася ВР (95% ДІ) на рівні 0,78 (0,68-0,90) [медіана ЗВ (95% ДІ) становила 11,9 місяців (10,88-13,01) для комбінації плацебо/FOLFIRI і 13,8 місяців для комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI (12,68-15,44)]
Інші аналізи підгруп з оцінкою загальної виживаності та виживаності без прогресування захворювання в залежності від віку (<65; ≥65), статі, наявності метастазів лише печінки, артеріальна гіпертензія у анамнезі та кількість уражених захворюванням органів продемонстрували більш високу ефективність лікування на фоні отримання комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI у порівнянні із комбінацією плацебо/FOLFIRI.
Аналіз загальної виживаності в підгрупах показав, що переваги лікування, порівнянні із такими в загальній популяції, на фоні отримання комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI мають пацієнти як віком <65 років, так і пацієнти віком ≥65 років.
Педіатрична популяція. Європейське агентство лікарських засобів звільнило виробника від зобов’язання щодо проведення досліджень з вивчення препарату ЗАЛТРАП (ZALTRAP) у всіх підгрупах педіатричної популяції з аденокарциномою товстого кишечнику та прямої кишки (інформація щодо застосування цього засобу у дітей наведена в розділі 4.2).
Фармакокінетичні властивості.
Описані нижче фармакокінетичні властивості препарату були у значній мірі визначені у популяційному фармакокінетичному аналізі за даними 1507 пацієнтів з різними типами злоякісних захворювань у пізніх стадіях.
Абсорбція. За даними доклінічних досліджень з використанням моделей пухлин біологічно активні дози афліберсепту корелювали з дозами, необхідними для досягнення циркулюючих концентрацій вільного афліберсепту, які б перевищували концентрацію афліберсепту, зв'язаного з VEGF. Циркулюючі концентрації афліберсепту, зв'язаного з VEGF, зростають зі збільшенням дози афліберсепту, поки більша частина доступного VEGF не буде зв'язаною. Подальше збільшення дози афліберсепту призводило до дозозалежного зростання концентрацій вільного афліберсепту в плазмі крові і лише невеликого додаткового зростання концентрації афліберсепту, зв'язаного з VEGF.
При застосуванні у пацієнтів ЗАЛТРАП (ZALTRAP) вводять в дозі 4 мг/кг маси тіла внутрішньовенно кожні два тижні, завдяки чому в організмі пацієнта концентрація циркулюючого вільного афліберсепту перевищує концентрацію афліберсепту, зв'язаного з VEGF.
При застосуванні препарату у рекомендованих дозах, тобто 4 мг/кг маси тіла кожні два тижні, концентрація вільного афліберсепту досягала рівнів, близьких до рівноважного стану, на початок другого циклу хіміотерапії, практично без кумуляції (коефіцієнт кумуляції становить 1,2 в рівноважному стані відносно концентрації після першого введення препарату).
Розподіл. Об'єм розподілу вільного афліберсепту в рівноважному стані становить приблизно 8 літрів.
Біотрансформація. Оскільки афліберсепт є білком, досліджень з вивчення його метаболізму не проводилося. Очікується, що афліберсепт розщеплюватиметься до низькомолекулярних пептидів та окремих амінокислот.
Елімінація. Вільний афліберсепт виводиться переважно через зв'язування з ендогенним VEGF з утворенням стабільного неактивного комплексу. Очікується, що, як і інші високомолекулярні білки, як вільний, так і зв'язаний афліберсепт буде повільніше виводитися в ході інших біологічних процесів, таких як протеолітична дисиміляція.
У дозах понад 2 мг/кг кліренс вільного афліберсепту становив 1,0 л/добу, а термінальний період напіввиведення становив 6 діб.
Високомолекулярні білки не виводяться нирковим шляхом, тому очікується, що ниркове виведення афліберсепту буде мінімальним.
Лінійність/Нелінійність. Відповідно тому, що розподіл даного лікарського засобу є опосередкованим мішенню, вільний афліберсепт демонструє більш швидкий (нелінійний) кліренс при дозах менш ніж 2 мг/кг – ймовірно, через високоафінне зв'язування афліберсепту з ендогенним VEGF. Лінійний кліренс, який спостерігався в діапазоні доз від 2 до 9 мг/кг, обумовлений, вірогідно, ненасичуваними біологічними механізмами виведення, такими як дисиміляція білків.
Інші окремі підгрупи пацієнтів.
Люди похилого віку. Вік пацієнта не впливає на фармакокінетику вільного афліберсепту.
Раса. У популяційному аналізі не було виявлено ніякого впливу расової приналежності на фармакокінетику препарату.
Стать. Стать була найбільш значущою коваріатою для пояснення міжіндивідуальної варіабельності вільного афліберсепту зі збільшенням кліренсу на 15,5% і об’єму розподілу на 20,6% у чоловіків у порівнянні із жінками. Завдяки дозуванню на основі маси тіла пацієнта ці відмінності не впливають на експозицію препарату, і корекція його дози з огляду на стать пацієнтів не потрібна.
Маса тіла. Маса тіла пацієнта впливала на кліренс та об'єм розподілу вільного афліберсепту, обумовлюючи зростання експозиції афліберсепту на 29% у пацієнтів з масою тіла ≥100 кг.
Печінкова дисфункція. Спеціальних досліджень з вивчення застосування препарату ЗАЛТРАП (ZALTRAP) у пацієнтів з порушенням функції печінки не проводилося. За результатами популяційного фармакокінетичного аналізу на матеріалі даних 1507 пацієнтів з різними видами злоякісних новоутворень на пізніх стадіях, які отримували ЗАЛТРАП (ZALTRAP) з хіміотерапією або без неї, ЗАЛТРАП (ZALTRAP) приймали 63 пацієнти з легким порушенням функції печінки (рівень загального білірубіну >1,0 × - 1,5 × верхньої межі норми [ВМН] і будь-який рівень АСТ) та 5 пацієнтів з помірним порушенням функції печінки (рівень загального білірубіну >1,5 × – 3 × ВМН і будь-який рівень АСТ). У цих пацієнтів з легким та помірним порушенням функції печінки це порушення не впливало на кліренс афліберсепту. Наразі немає даних щодо застосування препарату ЗАЛТРАП (ZALTRAP) у пацієнтів з тяжким порушенням функції печінки (рівень загального білірубіну >3 × ВМН і будь-який рівень АСТ).
Ниркова дисфункція. Спеціальних досліджень з вивчення застосування препарату ЗАЛТРАП (ZALTRAP) у пацієнтів з порушенням функції нирок не проводилося. Був проведений популяційний фармакокінетичний аналіз на матеріалі даних 1507 пацієнтів з різними видами злоякісних новоутворень на пізніх стадіях, які отримували ЗАЛТРАП (ZALTRAP) з хіміотерапією або без неї. До цієї популяції входили: 549 пацієнтів з легким порушенням функції нирок (кліренс креатиніну [CLCR] в діапазоні 50-80 мл/хв), 96 пацієнтів з помірним порушенням функції нирок (CLCR в діапазоні 30-50 мл/хв) та 5 пацієнтів з тяжким порушенням функції нирок (CLCR <30 мл/хв). За результатами цього популяційного фармакокінетичного аналізу не було виявлено жодних клінічно значущих змін кліренсу або системної експозиції (AUC) вільного афліберсепту у пацієнтів з помірним та легким порушенням функції нирок, які отримували ЗАЛТРАП (ZALTRAP) в дозі 4 мг/кг маси тіла, у порівнянні із загальною популяцією учасників дослідження. У зв’язку із дуже обмеженою кількістю даних не можна зробити жодних висновків щодо пацієнтів з тяжким порушенням функції нирок. У малочисельних пацієнтів з тяжким порушенням функції нирок експозиція препарату була подібною до такої, що спостерігалася у пацієнтів з нормальною функцією нирок.
Показання для застосування
ЗАЛТРАП (ZALTRAP) у комбінації з хіміотерапією іринотеканом/5-фторурацилом/фоліновою кислотою (схема FOLFIRI) показаний дорослим пацієнтам з метастатичним колоректальним раком (МКРР), який виявився резистентним до схеми хіміотерапії на основі оксаліплатину або прогресував на фоні її застосування.
Спосіб застосування і дози
ЗАЛТРАП (ZALTRAP) має застосовуватися під наглядом лікаря, що має досвід призначення протипухлинних препаратів.
Рекомендована доза препарату ЗАЛТРАП (ZALTRAP), який вводять шляхом внутрішньовенної інфузії протягом 1 години, становить 4 мг/кг маси тіла, з подальшим застосуванням схеми FOLFIRI. Це вважається одним циклом лікування.
Схема FOLFIRI, що має використовуватися при цьому, включає одночасне введення у день 1 за допомогою системи для внутрішньовенних інфузій з Y-подібним конектором іринотекану 180 мг/м2 у вигляді внутрішньовенної інфузії протягом 90 хвилин та фолінової кислоти (рацемічна суміш право- та лівообертальних ізомерів) 400 мг/м2 у вигляді внутрішньовенної інфузії протягом 2 годин, з подальшим введенням 5-фторурацилу (5-ФУ) 400 мг/м2 у вигляді внутрішньовенного болюсу, а потім – 5-ФУ 2400 мг/м2 у вигляді безперервної внутрішньовенної інфузії протягом 46 годин. Цей цикл лікування повторюється кожні 2 тижні.
Лікування препаратом ЗАЛТРАП (ZALTRAP) має продовжуватися, поки не буде виявлене прогресування захворювання або поки не виникнуть неприйнятні токсичні реакції.
Модифікація дози. Застосування препарату ЗАЛТРАП (ZALTRAP) необхідно відмінити в наступних випадках (див. розділ «Особливості застосування»):
- тяжкі геморагічні явища;
- перфорація органів шлунково-кишкового тракту (ШКТ);
- утворення нориць;
- артеріальна гіпертензія, що не контролюється належним чином антигіпертензивною терапією, або розвиток гіпертензивного кризу чи гіпертензивної енцефалопатії;
- артеріальні тромбоемболічні події (АТЕ);
- венозні тромбоемболічні події 4 ступеня тяжкості (в тому числі тромбоемболія легеневої артерії);
- нефротичний синдром або тромботична мікроангіопатія (ТМА);
- тяжкі реакції гіперчутливості (в тому числі бронхоспазм, задишка, ангіоневротичний набряк та анафілаксія) (див. розділи «Протипоказання» та «Особливості застосування»);
- порушення загоєння ран, яке потребує медичного втручання
- синдром оборотної задньої енцефалопатії (СОЗЕ) (також відомий як синдром оборотної задньої лейкоенцефалопатії (СОЗЛ))
Прийом препарату ЗАЛТРАП (ZALTRAP) слід тимчасово припинити щонайменше за 4 тижні до планової хірургічної операції (див. розділ «Особливості застосування»).
|
Тимчасове припинення застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI або модифікація доз |
||
|
Нейтропенія або тромбоцитопенія (див. розділи «Особливості застосування» та «Побічні реакції») |
Застосування засобів ЗАЛТРАП (ZALTRAP)/FOLFIRI слід тимчасово припинити, поки кількість нейтрофілів не становитиме ≥1,5 × 109/л або поки кількість тромбоцитів не становитиме ≥75 × 109/л. |
|
|
Фебрильна нейтропенія або нейтропенічний сепсис |
Зменшити дозу іринотекану на 15-20% в наступних циклах хіміотерапії. При повторних проявах – додатково зменшити дози 5-ФУ для болюсного та інфузійного введення на 20% в наступних циклах. При повторних проявах після зменшення доз іринотекану та 5-ФУ слід зважити доцільність зниження дози препарату ЗАЛТРАП (ZALTRAP) до 2 мг/кг. Може розглядатися можливість застосування гранулоцитарного колонієстимулюючого фактору (Г-КСФ). |
|
|
Легкі або середньоважкі реакції гіперчутливості на препарат ЗАЛТРАП (ZALTRAP) (в тому числі гіперемія, висипання, кропивниця та свербіння) (див. розділ «Особливості застосування») |
Інфузію слід тимчасово припинити, поки реакція гіперчутливості не зникне. За наявності клінічних показань можна призначати кортикостероїди та/або антигістамінні засоби. У наступних циклах хіміотерапії може бути розглянута можливість попереднього застосування кортикостероїдів та/або антигістамінних засобів. |
|
|
Тяжкі реакції гіперчутливості (в тому числі бронхоспазм, задишка, ангіоневротичний набряк та анафілаксія) (див. розділи «Протипоказання» та «Особливості застосування») |
Припинити застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI та призначити відповідну медикаментозну терапію. |
|
|
Тимчасове припинення застосування препарату ЗАЛТРАП (ZALTRAP) та модифікація дози |
||
|
Артеріальна гіпертензія (див. розділ «Особливості застосування») |
Застосування препарату ЗАЛТРАП (ZALTRAP) слід тимчасово припинити, поки не буде досягнутий контроль над артеріальною гіпертензією. При повторних проявах тяжкої артеріальної гіпертензії застосування препарату слід тимчасово припинити, поки не буде досягнутий контроль над гіпертензією, і в наступних циклах хіміотерапії зменшити дозу препарату до 2 мг/кг. |
|
|
Протеїнурія (див. розділ «Особливості застосування») |
Слід тимчасово припинити застосування препарату ЗАЛТРАП (ZALTRAP), якщо рівень екскреції білка з сечею становить ≥2 г/24 год, та поновити введення препарату, коли рівень екскреції білка з сечею становитиме <2 г/24 год. При повторних проявах протеїнурії слід тимчасово припинити застосування препарату, поки рівень білка в сечі не зменшиться до <2 г/добу, після чого зменшити дозу препарату до 2 мг/кг. |
|
|
Модифікація доз препаратів схеми FOLFIRI при її застосуванні в комбінації з препаратом ЗАЛТРАП (ZALTRAP) |
||
|
Тяжкий стоматит та синдром долонно-підошовної еритродизестезії |
Слід зменшити дози 5-ФУ для болюсного та інфузійного введення на 20%. |
|
|
Тяжка діарея |
Слід зменшити дозу іринотекану на 15-20%. Якщо тяжка діарея повторюється на наступному циклі хіміотерапії, слід також зменшити дози 5-ФУ для болюсного та інфузійного введення на 20%. Якщо тяжка діарея не припиняється і після зменшення дози обох цих препаратів, застосування схеми FOLFIRI слід припинити. За необхідності можна застосовувати протидіарейні лікарські засоби та засоби для регідратації. |
|
При виникненні інших токсичних реакцій, пов'язаних із застосуванням іринотекану, 5-ФУ або фолінової кислоти, слід керуватися відповідними чинними Загальними характеристиками лікарських засобів.
Особливі групи пацієнтів
Люди похилого віку. У базовому дослідженні за участі пацієнтів з МКРР 28,2% пацієнтів були віком ≥65 і <75 років, і 5,4% пацієнтів були віком ≥75 років. У пацієнтів похилого віку корекція дози препарату ЗАЛТРАП (ZALTRAP) не потрібна.
Печінкова дисфункція. Спеціальних досліджень з вивчення застосування препарату ЗАЛТРАП (ZALTRAP) у пацієнтів з порушенням функції печінки не проводилося (див. розділ «Фармакокінетичні властивості»). Клінічні дані свідчать про те, що у пацієнтів з легкою або помірною печінковою дисфункцією зміна дози афліберсепту не потрібна. Дані із застосування афліберсепту у пацієнтів з тяжкою печінковою дисфункцією наразі відсутні.
Ниркова дисфункція. Спеціальних досліджень з вивчення застосування препарату ЗАЛТРАП (ZALTRAP) у пацієнтів з порушенням функції нирок не проводилося (див. розділ «Фармакокінетичні властивості»). Клінічні дані свідчать про те, що у пацієнтів з легкою або помірною нирковою дисфункцією зміна початкової дози препарату не потрібна. Дані із застосування препарату у пацієнтів з тяжкою нирковою дисфункцією наразі дуже обмежені, тому лікування таких пацієнтів має проводитися з обережністю.
Педіатрична популяція. Релевантний досвід застосування препарату ЗАЛТРАП (ZALTRAP) в педіатричній популяції за таким показанням, як метастатичний колоректальний рак, відсутній.
Спосіб застосування.
ЗАЛТРАП (ZALTRAP) показаний для застосування тільки у вигляді внутрішньовенної інфузії протягом 1 години. Через гіперосмоляльність (1000 мОсм/кг) концентрату ЗАЛТРАП (ZALTRAP) нерозведений концентрат препарату ЗАЛТРАП (ZALTRAP) не можна вводити внутрішньовенно струменево або болюсно. ЗАЛТРАП (ZALTRAP) не можна вводити інтравітреально (див. розділи «Протипоказання» та «Особливості застосування»).
Кожен флакон концентрату для приготування розчину для інфузій призначений лише для однократного використання (для введення однієї дози препарату).
Розведений розчин препарату ЗАЛТРАП (ZALTRAP) слід вводити за допомогою системи для інфузій з поліефірсульфоновим фільтром з діаметром пор 0,2 мікрони.
Інфузійна система має бути вироблена з одного з наступних матеріалів:
- полівінілхлорид (ПВХ), що містить ди(2-етилгексил)фталат (ДЕГФ);
- ПВХ без вмісту ДЕГФ, з триоктилтримелітатом (TOTM);
- поліпропілен;
- вистелений поліетиленом ПВХ;
- поліуретан.
Не можна використовувати фільтри з полівініліденфториду (ПВДФ) або нейлону.
Запобіжні заходи, що мають бути застосовані перед початком підготовки препарату до введення або перед введенням препарату
ЗАЛТРАП (ZALTRAP) – стерильний апірогенний концентрат без консервантів, тому приготування розчину для інфузій має виконуватися медичним працівником, із дотриманням правил техніки безпеки, в асептичних умовах.
При застосуванні препарату ЗАЛТРАП (ZALTRAP) слід дотримуватися обережності, що включає використання захисного обладнання, засобів особистого захисту (наприклад, рукавичок) та спеціальних підготовчих процедур.
Приготування інфузійного розчину.
- Перед використанням оглянути флакон з препаратом ЗАЛТРАП (ZALTRAP). Концентрат має бути прозорим, без сторонніх часточок.
- У залежності від необхідної пацієнтові дози відібрати з флакону потрібну кількість концентрату ЗАЛТРАП (ZALTRAP). Для приготування розчину для інфузій може знадобитися більше одного флакона.
- Розвести концентрат до необхідного для введення об'єму, додавши розчин хлориду натрію 9 мг/мл (0,9%) або 5% розчин глюкози для інфузій. Концентрація готового для внутрішньовенної інфузії розчину препарату ЗАЛТРАП (ZALTRAP) має знаходитися в межах 0,6–8 мг/мл афліберсепту.
- Використовувати інфузійні пакети, вироблені з ПВХ з вмістом ДЕГФ або з поліолефінів.
- Розведений розчин перед введенням необхідно оглянути на наявність твердих часточок або зміни кольору. За наявності будь-яких змін кольору розчину або включень твердих часточок розведений розчин використовувати не можна.
- ЗАЛТРАП (ZALTRAP) – флакон для разового використання. Не можна повторно відбирати вміст із флакона після першого проколу пробки. Будь-яка невикористана кількість концентрату має бути утилізована.
Утилізація.
Всі невикористані рештки лікарського засобу та інші відходи необхідно утилізувати у відповідності до місцевих вимог.
Побічні реакції
Загальна характеристика профілю безпечності препарату. Безпека препарату ЗАЛТРАП (ZALTRAP) у комбінації зі схемою FOLFIRI оцінювалася у дослідженні III фази за участю 1216 пацієнтів, які раніше отримували лікування з приводу метастатичного колоректального раку, з яких 611 пацієнтів приймали ЗАЛТРАП (ZALTRAP) 4 мг/кг в комбінації зі схемою FOLFIRI кожні 2 тижні (один цикл) і 605 пацієнтів отримували комбінацію плацебо/FOLFIRI. Медіана кількості отриманих циклів лікування з використанням комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI становила 9.
Найбільш частими побічними реакціями (усіх ступенів тяжкості, з частотою виникнення ≥20%) в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI, які зустрічалися щонайменше на 2% частіше, ніж в групі застосування плацебо/FOLFIRI, були такі (в порядку зменшення частоти виникнення): лейкопенія, діарея, нейтропенія, протеїнурія, підвищення рівня аспартатамінотрансферази (АСТ), стоматит, підвищена втомлюваність, тромбоцитопенія, підвищення рівня аланінамінотрансферази (АЛТ), артеріальна гіпертензія, зменшення маси тіла, зниження апетиту, носова кровотеча, біль у животі, дисфонія, підвищення рівня креатиніну в сироватці крові і головний біль (див. Таблицю 1).
Найбільш частими побічними реакціями 3-4 ступенів тяжкості (з частотою виникнення ≥5%) в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI, які зустрічалися щонайменше на 2% частіше, ніж в групі застосування плацебо/FOLFIRI, були такі (в порядку зменшення частоти виникнення): нейтропенія, діарея, артеріальна гіпертензія, лейкопенія, стоматит, підвищена втомлюваність, протеїнурія та загальна слабкість (див. Таблицю 1).
Найбільш частими побічними реакціями, які обумовили необхідність повної відміни комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI у ≥1% пацієнтів, були судинні порушення (3,8%), в тому числі артеріальна гіпертензія (2,3%), інфекції (3,4%), загальна слабкість/підвищена втомлюваність (1,6%, 2,1%), діарея (2,3%), дегідратація (1%), стоматит (1,1%), нейтропенія (1,1%), протеїнурія (1,5%) та тромбоемболія легеневої артерії (1,1%).
Табличний перелік побічними реакцій. У Таблиці 1 наведений перелік побічних реакцій та відхилень від норми з боку результатів лабораторних аналізів, що спостерігалися у пацієнтів, які отримували комбінацію ЗАЛТРАП (ZALTRAP)/FOLFIRI, в порівнянні з пацієнтами, які отримували плацебо/ЗАЛТРАП (ZALTRAP), згрупованих за категоріями «Система-Орган-Клас» за класифікацією MedDRA та за частотою виникнення. Побічні реакції, представлені у Таблиці 1, визначаються як будь-які клінічні реакції або відхилення від норми з боку результатів лабораторних аналізів (всіх ступенів тяжкості), що зустрічалися на ≥2% частіше у пацієнтів в групі прийому афліберсепту у порівнянні із групою плацебо у дослідженні за участю пацієнтів з МКРР; в таблицю включені також ті реакції, які не досягли цього порогового значення різниці частоти, але узгоджуються із класовими ефектами анти-VEGF препаратів і спостерігалися у будь-якому дослідженні з вивчення афліберсепту. Інтенсивність побічних реакцій класифікована відповідно до NCI CTC (Загальні критерії токсичності Національного онкологічного інституту США), версія 3.0 (ступінь тяжкості ≥3 = G≥3). Частота побічних реакцій визначалася для усіх ступенів тяжкості з використанням таких критеріїв: дуже часто (≥1/10); часто (від ≥ 1/100 до < 1/10); нечасто (від ≥ 1/1000 до < 1/100); рідко (від ≥ 1/10 000 до < 1/1000); дуже рідко (< 1/10 000); частота невідома (неможливо оцінити за доступними даними).
Таблиця 1. Побічні реакції, які зустрічалися у пацієнтів на фоні застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI в дослідженні за участю пацієнтів з МКРР
|
Система-Орган-Клас Категорія частоти |
Побічні реакції |
|
||
|
Всі ступені тяжкості |
Ступені тяжкості ≥3 |
|
||
|
Інфекційні та паразитарні захворювання |
|
|||
|
Дуже часто |
Інфекція (1) |
Інфекція (1) |
|
|
|
Часто |
Нейтропенічна інфекція/сепсис (1) Інфекція сечових шляхів Назофарингіт |
Нейтропенічна інфекція/сепсис (1) |
|
|
|
Нечасто |
Інфекція сечових шляхів |
|
||
|
Розлади з боку крові та лімфатичної системи |
|
|||
|
Дуже часто |
Лейкопенія (2) Нейтропенія (1), (2) Тромбоцитопенія (2) |
Лейкопенія (2) Нейтропенія (2) |
|
|
|
Часто |
Фебрильна нейтропенія |
Фебрильна нейтропенія Тромбоцитопенія (2) |
|
|
|
Розлади з боку імунної системи |
|
|||
|
Часто |
Гіперчутливість (1) |
|
||
|
Нечасто |
Гіперчутливість (1) |
|||
|
Метаболічні та аліментарні розлади |
||||
|
Дуже часто |
Погіршення апетиту Зниження маси тіла |
|||
|
Часто |
Дегідратація (1) |
Дегідратація (1) Погіршення апетиту Зниження маси тіла |
||
|
Розлади з боку нервової системи |
||||
|
Дуже часто |
Головний біль |
|||
|
Часто |
Головний біль |
|||
|
Нечасто |
СОЗЕ (1), (4) |
СОЗЕ (1), (4) |
||
|
Розлади з боку судин |
||||
|
Дуже часто |
Артеріальна гіпертензія (1) Геморагічне явище (1) |
Артеріальна гіпертензія |
||
|
Часто |
Артеріальний тромбоемболізм (1) Венозний тромбоемболізм (1) |
Артеріальний тромбоемболізм (1) Венозний тромбоемболізм (1) Геморагічне явище (1) |
||
|
Розлади з боку респіраторної системи, грудної клітки та середостіння |
||||
|
Дуже часто |
Задишка Носова кровотеча Дисфонія |
|||
|
Часто |
Біль у ротоглотці Ринорея |
|||
|
Нечасто |
Задишка Носова кровотеча Дисфонія Біль у ротоглотці |
|||
|
Розлади з боку шлунково-кишкової системи |
||||
|
Дуже часто |
Діарея (1) Стоматит Біль в животі Біль у верхніх відділах живота |
Діарея (1) Стоматит |
||
|
Часто |
Ректальна кровотеча Нориця (1) Афтозний стоматит Геморой Прокталгія Зубний біль |
Біль в животі Біль у верхніх відділах живота |
||
|
Нечасто |
Перфорація органів ШКТ (1) |
Перфорація органів ШКТ (1) Ректальна кровотеча Нориця (1) Афтозний стоматит Прокталгія |
|
|
|
Гепатобіліарні розлади |
||||
|
Дуже часто |
Підвищений рівень АСТ (2) Підвищений рівень АЛТ (2) |
|
||
|
Часто |
Підвищений рівень АСТ (2) Підвищений рівень АЛТ (2) |
|
||
|
Розлади з боку шкіри та підшкірної клітковини |
||||
|
Дуже часто |
Синдром долонно-підошовної еритродизестезії |
|
||
|
Часто |
Гіперпігментація шкіри |
Синдром долонно-підошовної еритродизестезії |
|
|
|
Нечасто |
Порушення загоєння ран (1) |
Порушення загоєння ран (1) |
|
|
|
Розлади з боку нирок та сечовивідного тракту |
||||
|
Дуже часто |
Протеїнурія (1), (3) Підвищення рівня сироваткового креатиніну |
|
||
|
Часто |
Протеїнурія (1), (3) |
|
||
|
Нечасто |
Нефротичний синдром (1) Тромботична мікроангіопатія (1) |
Нефротичний синдром (1) Тромботична мікроангіопатія (1) |
|
|
|
Загальні розлади та реакції у місці введення |
||||
|
Дуже часто |
Астенічні стани |
Астенічні стани |
|
|
|
Примітка: Побічні реакції реєструвалися з використанням Медичного словника нормативно-правової діяльності (MedDRA) версії MEDDRA 13.1 і оцінювалися за ступенем тяжкості за Загальними критеріями токсичності Національного онкологічного інституту США (NCI CTC) версії 3.0 (1) Див. параграф «Опис окремих небажаних реакцій» у цьому розділі (2) На основі лабораторних показників (відсотки розраховувалися від числа пацієнтів, у яких виконувалися лабораторні оцінки) (3) Компіляція клінічних та лабораторних даних (4) Не були зареєстровані у дослідженні за участю пацієнтів з МКРР; проте СОЗЕ спостерігався у пацієнтів, що брали участь в інших дослідженнях, де вони отримували афліберсепт у якості монотерапії або у комбінації з хіміотерапевтичними засобами, відмінними від засобів схеми FOLFIRI |
||||
У базовому дослідженні за участю пацієнтів з МКРР такі побічні явища, як анемія, нудота, блювання, запор, алопеція, підвищення рівня лужної фосфатази та гіпербілірубінемія, спостерігалися у ≥20% пацієнтів. Частота цих небажаних реакцій була порівнянною в обох групах, і різниця частоти в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI у порівнянні із групою плацебо не перевищувала поріг ≥2%.
Опис окремих побічних реакцій.
Геморагічні явища. У пацієнтів, які отримують ЗАЛТРАП (ZALTRAP), підвищується ризик виникнення геморагічних явищ, в тому числі тяжких і іноді фатальних. У базовому дослідженні за участю пацієнтів з МКРР випадки виникнення кровотеч/крововиливів (усіх ступенів тяжкості) спостерігалися у 37,8% пацієнтів, які приймали ЗАЛТРАП (ZALTRAP)/FOLFIRI, порівняно з 19,0% пацієнтів, які отримували плацебо/FOLFIRI. Найбільш часто спостерігалася невелика (1-2 ступеня) носова кровотеча, яка зустрічалася у 27,7% пацієнтів, що отримували комбінацію ЗАЛТРАП (ZALTRAP)/FOLFIRI. Геморагічні явища 3-4 ступенів тяжкості, серед яких ШК-кровотечі, гематурія та післяпроцедурні геморагічні явища, були зареєстровані у 2,9% пацієнтів, які приймали ЗАЛТРАП (ZALTRAP)/FOLFIRI, порівняно з 1,7% пацієнтів, які отримували плацебо/FOLFIRI. В інших дослідженнях у пацієнтів, які приймали ЗАЛТРАП (ZALTRAP), спостерігалися тяжкі внутрішньочерепні крововиливи та легеневі кровотечі/кровохаркання, в тому числі фатальні (див. розділ «Особливості застосування»).
Перфорація органів шлунково-кишкового тракту. У пацієнтів, які приймали ЗАЛТРАП (ZALTRAP), спостерігалися випадки виникнення перфорації органів ШКТ, в тому числі фатальні. У базовому дослідженні за участю пацієнтів з МКРР випадки виникнення перфорації органів ШКТ (усіх ступенів тяжкості) спостерігалися у 3 з 611 пацієнтів (0,5%), які приймали ЗАЛТРАП (ZALTRAP)/FOLFIRI, порівняно з 3 з 605 пацієнтів (0,5%), які отримували плацебо/FOLFIRI. Перфорації органів ШКТ 3-4 ступенів тяжкості спостерігалися в усіх 3 пацієнтів (0,5%) в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI та у 2 пацієнтів (0,3%) в групі застосування плацебо/FOLFIRI. За результатами трьох плацебо-контрольованих клінічних досліджень ІІІ фази (за участю пацієнтів з колоректальним раком, раком підшлункової залози та раком легень) частота виникнення перфорації органів ШКТ (усіх ступенів тяжкості) становила 0,8% у пацієнтів, які приймали ЗАЛТРАП (ZALTRAP), та 0,3% у пацієнтів, які отримували плацебо. Перфорації органів ШКТ 3-4 ступенів тяжкості спостерігалися у 0,8% пацієнтів, які приймали ЗАЛТРАП (ZALTRAP), та у 0,2% пацієнтів, які отримували плацебо (див. розділ «Особливості застосування»).
Утворення нориць. У пацієнтів, які отримували ЗАЛТРАП (ZALTRAP), спостерігалися випадки утворення нориць, як в ШКТ, так і поза ШКТ. У базовому дослідженні за участю пацієнтів з МКРР випадки виникнення нориць (анальних, тонкокишково-сечоміхурних, тонкокишково-шкірних, товстокишково-піхвових, кишкових) спостерігалися у 9 з 611 пацієнтів (1,5%) в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI та у 3 з 605 пацієнтів (0,5%) в групі застосування плацебо/FOLFIRI. Утворення ШК-нориць 3 ступеня тяжкості спостерігалося у 2 пацієнтів, які приймали ЗАЛТРАП (ZALTRAP) (0,3%), та у 1 пацієнта, який отримував плацебо (0,2%). За результатами трьох плацебо-контрольованих клінічних досліджень ІІІ фази (за участю пацієнтів з колоректальним раком, раком підшлункової залози та раком легень) частота виникнення нориць (усіх ступенів тяжкості) становила 1,1% у пацієнтів, які приймали ЗАЛТРАП (ZALTRAP), та 0,2% у пацієнтів, які отримували плацебо. Нориці 3-4 ступенів тяжкості спостерігалися у 0,2% пацієнтів, які приймали ЗАЛТРАП (ZALTRAP), та у 0,1% пацієнтів, які отримували плацебо (див. розділ «Особливості застосування»).
Артеріальна гіпертензія. У базовому дослідженні за участю пацієнтів з МКРР артеріальна гіпертензія (усіх ступенів тяжкості) спостерігалася у 41,2% пацієнтів у групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI та у 10,7% пацієнтів у групі застосування плацебо/FOLFIRI. У пацієнтів, які отримували комбінацію ЗАЛТРАП (ZALTRAP)/FOLFIRI, спостерігався підвищений ризик артеріальної гіпертензії 3-4 ступеня тяжкості (в тому числі артеріальна гіпертензія та один випадок есенціальної гіпертензії). Артеріальна гіпертензія 3 ступеня тяжкості (яка потребувала корекції існуючої антигіпертензивної терапії або застосування більше ніж одного лікарського засобу) виникла у 1,5% пацієнтів у групі застосування плацебо/FOLFIRI та у 19,1% пацієнтів у групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI. Артеріальна гіпертензія 4 ступеня тяжкості (гіпертензивний криз) була зареєстрована у 1 пацієнта (0,2%) в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI. Серед цих пацієнтів, які отримували комбінацію ЗАЛТРАП (ZALTRAP)/FOLFIRI і у яких розвинулася артеріальна гіпертензія 3-4 ступенів тяжкості, у 54% захворювання виникло під час перших двох циклів лікування (див. розділ «Особливості застосування»).
Тромботичні та емболічні явища.
Артеріальні тромбоемболічні події. У базовому дослідженні за участю пацієнтів з МКРР АТЕ (включаючи транзиторні ішемічні атаки, цереброваскулярні події, стенокардію, внутрішньосерцевий тромб, інфаркт міокарда, артеріальну тромбоемболію та ішемічний коліт) спостерігалися у 2,6% пацієнтів у групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI та у 1,5% пацієнтів у групі застосування плацебо/FOLFIRI. Події 3-4 ступенів тяжкості спостерігалися у 11 пацієнтів (1,8%) в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI та у 3 пацієнтів (0,5%) в групі застосування плацебо/FOLFIRI. За результатами трьох плацебо-контрольованих клінічних досліджень ІІІ фази (за участю пацієнтів з колоректальним раком, раком підшлункової залози та раком легень) частота виникнення АТЕ (усіх ступенів тяжкості) становила 2,3% у пацієнтів, які приймали ЗАЛТРАП (ZALTRAP), та 1,7% у пацієнтів, які отримували плацебо. АТЕ 3-4 ступенів тяжкості спостерігалися у 1,7% пацієнтів, які приймали ЗАЛТРАП (ZALTRAP), та у 1,0% пацієнтів, які отримували плацебо (див. розділ «Особливості застосування»).
Венозні тромбоемболічні події. До венозних тромбоемболічних подій (ВТЕ) відносилися тромбоз глибоких вен і тромбоемболія легеневої артерії. У базовому дослідженні за участю пацієнтів з МКРР випадки виникнення ВТЕ усіх ступенів тяжкості спостерігалися у 9,3% пацієнтів, які приймали ЗАЛТРАП (ZALTRAP)/FOLFIRI, порівняно з 7,3% пацієнтів, які отримували плацебо/FOLFIRI. ВТЕ 3-4 ступенів тяжкості спостерігалися у 7,9% пацієнтів в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI та у 6,3% пацієнтів в групі застосування плацебо/FOLFIRI. Випадки тромбоемболії легеневої артерії спостерігалися у 4,6% пацієнтів в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI та у 3,5% пацієнтів в групі застосування плацебо/FOLFIRI. За результатами трьох плацебо-контрольованих клінічних досліджень ІІІ фази (за участю пацієнтів з колоректальним раком, раком підшлункової залози та раком легень) частота виникнення ВТЕ (усіх ступенів тяжкості) становила 7,1% у пацієнтів, які приймали ЗАЛТРАП (ZALTRAP), та 7,1% у пацієнтів, які отримували плацебо.
Протеїнурія. У базовому дослідженні за участю пацієнтів з МКРР випадки виникнення протеїнурії (за зведеними клінічними і лабораторними даними) спостерігалися у 62,2% пацієнтів, які приймали ЗАЛТРАП (ZALTRAP)/FOLFIRI, порівняно з 40,7% пацієнтів, які отримували плацебо/FOLFIRI. Протеїнурія 3-4 ступенів тяжкості спостерігалася у 7,9% пацієнтів в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI порівняно з 1,2% пацієнтів в групі застосування плацебо/FOLFIRI. Нефротичний синдром був зареєстрований у 2 пацієнтів (0,5%) групи застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI і не виявлений у жодного з пацієнтів групи застосування плацебо/FOLFIRI. У одного пацієнта з групи ЗАЛТРАП (ZALTRAP)/FOLFIRI з існуючими протеїнурією та артеріальною гіпертензією розвинулася тромботична мікроангіопатія (ТМА). За результатами трьох плацебо-контрольованих клінічних досліджень ІІІ фази (за участю пацієнтів з колоректальним раком, раком підшлункової залози та раком легень) частота виникнення нефротичного синдрому становила 0,5% у пацієнтів, які приймали ЗАЛТРАП (ZALTRAP), та 0,1% у пацієнтів, які отримували плацебо (див. розділ «Особливості застосування»).
Нейтропенія та нейтропенічні ускладнення. У базовому дослідженні за участю пацієнтів з МКРР нейтропенія (усіх ступенів тяжкості) спостерігалася у 67,8% пацієнтів у групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI та у 56,3% пацієнтів у групі застосування плацебо/FOLFIRI. Нейтропенія 3-4 ступенів тяжкості спостерігалася у 36,7% пацієнтів в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI порівняно з 29,5% пацієнтів в групі застосування плацебо/FOLFIRI. Найбільш часто з нейтропенічних ускладнень 3-4 ступенів тяжкості виникала фебрильна нейтропенія, яка розвинулася у 4,3% пацієнтів у групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI порівняно з 1,7% пацієнтів у групі застосування плацебо/FOLFIRI. Нейтропенічна інфекція/сепсис 3-4 ступенів тяжкості спостерігалися у 1,5% пацієнтів в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI та у 1,2% пацієнтів в групі застосування плацебо/FOLFIRI (див. розділ «Особливості застосування»).
Інфекції. Інфекції більш часто виникали у пацієнтів, які приймали ЗАЛТРАП (ZALTRAP)/FOLFIRI (інфекції всіх ступенів тяжкості – 46,2%; 3-4 ступенів тяжкості – 12,3%), ніж у пацієнтів, які отримували плацебо/FOLFIRI (інфекції всіх ступенів тяжкості – 32,7%; 3-4 ступенів тяжкості – 6,9%). До них належали інфекція сечовивідних шляхів, назофарингіт, інфекція верхніх дихальних шляхів, пневмонія, інфекції в місці введення катетера та інфекції зубів.
Діарея та дегідратація. У базовому дослідженні за участю пацієнтів з МКРР діарея (усіх ступенів тяжкості) спостерігалася у 69,2% пацієнтів у групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI та у 56,5% пацієнтів у групі застосування плацебо/FOLFIRI. Дегідратація (всіх ступенів тяжкості) спостерігалася у 9,0% пацієнтів, які отримували ЗАЛТРАП (ZALTRAP)/FOLFIRI, та у 3,0% пацієнтів, які отримували плацебо/FOLFIRI. Діарея 3-4 ступенів тяжкості спостерігалася у 19,3% пацієнтів в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI порівняно з 7,8% пацієнтів в групі застосування плацебо/FOLFIRI. Дегідратація 3-4 ступенів тяжкості спостерігалася у 4,3% пацієнтів в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI порівняно з 1,3% пацієнтів в групі застосування плацебо/FOLFIRI (див. розділ «Особливості застосування»).
Реакції гіперчутливості. У базовому дослідженні за участю пацієнтів з МКРР тяжкі реакції гіперчутливості спостерігалися у 0,3% пацієнтів, які приймали ЗАЛТРАП (ZALTRAP)/FOLFIRI, порівняно з 0,5% пацієнтів, які отримували плацебо/FOLFIRI (див. розділ «Особливості застосування»).
Порушення загоєння ран. Застосування препарату ЗАЛТРАП (ZALTRAP) може призводити до порушення процесу загоєння ран (розходження країв рани, неспроможність анастомозу). У базовому дослідженні за участю пацієнтів з МКРР випадки порушення процесу загоєння ран спостерігалися у 3 пацієнтів (0,5%), які приймали ЗАЛТРАП (ZALTRAP)/FOLFIRI, порівняно з 5 пацієнтами (0,8%), які отримували плацебо/FOLFIRI. Порушення загоєння ран 3 ступеня тяжкості спостерігалося у 2 пацієнтів (0,3%) в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI і у жодного з пацієнтів в групі застосування плацебо/FOLFIRI. За результатами трьох плацебо-контрольованих клінічних досліджень ІІІ фази (за участю пацієнтів з колоректальним раком, раком підшлункової залози та раком легень) частота порушення процесу загоєння ран (усіх ступенів тяжкості) становила 0,5% у пацієнтів, які приймали ЗАЛТРАП (ZALTRAP), та 0,4% у пацієнтів, які отримували плацебо. Порушення загоєння ран 3-4 ступенів тяжкості спостерігалися у 0,2% пацієнтів, які приймали ЗАЛТРАП (ZALTRAP), та у жодного з пацієнтів, які отримували плацебо (див. розділ «Особливості застосування»).
Синдром зворотної задньої енцефалопатії (СЗЗЕ). У базовому дослідженні III фази за участю пацієнтів з МКРР випадків виникнення СЗЗЕ зареєстровано не було. У інших дослідженнях повідомлялося про випадки виникнення СЗЗЕ у пацієнтів, які отримували ЗАЛТРАП (ZALTRAP) у якості монотерапії (0,5%) і у комбінації з іншими хіміотерапевтичними засобами (див. розділ «Особливості застосування»).
Інші небажані реакції та відхилення від норми з боку результатів лабораторних аналізів (усіх ступенів тяжкості), частота виникнення яких відрізнялася на ≥5% у пацієнтів в групі ЗАЛТРАП (ZALTRAP)/FOLFIRI порівняно з групою пацієнтів, які отримували плацебо/FOLFIRI. Побічні реакції та відхилення від норми з боку результатів лабораторних аналізів (усіх ступенів тяжкості), частота виникнення яких відрізнялася на ≥5% у пацієнтів в групі ЗАЛТРАП (ZALTRAP)/FOLFIRI порівняно з групою пацієнтів, які отримували плацебо/FOLFIRI, були такими: лейкопенія (78,3% проти 72,4% усіх ступенів тяжкості; 15,6% проти 12,2% 3-4 ступенів тяжкості), підвищення рівня АСТ (57,5% проти 50,2% усіх ступенів тяжкості; 3,1% проти 1,7% 3-4 ступенів тяжкості), стоматит (50,1% проти 32,9% усіх ступенів тяжкості; 12,8% проти 4,6% 3-4 ступенів тяжкості), підвищена втомлюваність (47,8% проти 39,0% усіх ступенів тяжкості; 12,6% проти 7,8% 3-4 ступенів тяжкості), тромбоцитопенія (47,4% проти 33,8% усіх ступенів тяжкості; 3,3% проти 1,7% 3-4 ступенів тяжкості), підвищення рівня АЛТ (47,3% проти 37,1% усіх ступенів тяжкості; 2,7% проти 2,2% 3-4 ступенів тяжкості), порушення апетиту (31,9% проти 23,8% усіх ступенів тяжкості; 3,4% проти 1,8% 3-4 ступенів тяжкості), зменшення маси тіла (31,9% проти 14,4% усіх ступенів тяжкості; 2,6% проти 0,8% 3-4 ступенів тяжкості), дисфонія (25,4% проти 3,3% усіх ступенів тяжкості; 0,5% проти 0 3-4 ступенів тяжкості), головний біль (22,3% проти 8,8% усіх ступенів тяжкості; 1,6% проти 0,3% 3-4 ступенів тяжкості), загальна слабкість (18,3% проти 13,2% усіх ступенів тяжкості; 5,1% проти 3,0% 3-4 ступенів тяжкості), синдром долонно-підошовної еритродизестезії (11,0% проти 4,3% усіх ступенів тяжкості; 2,8% проти 0,5% 3-4 ступенів тяжкості) та гіперпігментація шкіри (8,2% проти 2,8% усіх ступенів тяжкості; 0 проти 0 3-4 ступенів тяжкості).
Педіатрична популяція. Безпечність цього лікарського засобу для педіатричної популяції наразі не встановлена.
Інші окремі підгрупи пацієнтів.
Люди похилого віку. У базовому дослідженні за участю пацієнтів з МКРР в групі застосування комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI 172 пацієнта (28,2%) були у віці ≥65 і <75 років, і 33 пацієнта (5,4%) – у віці ≥75 років. У пацієнтів похилого віку (≥65 років) може бути більш висока вірогідність розвитку небажаних реакцій. У пацієнтів похилого віку частота діареї, запаморочення, загальної слабкості, зниження маси тіла та дегідратації була на ≥5% вищою за таку у пацієнтів молодшого віку. Необхідно ретельно контролювати стан літніх пацієнтів щодо ознак розвитку діареї та можливої дегідратації (див. розділ «Особливості застосування»).
Ниркова дисфункціяє. У трьох плацебо-контрольованих клінічних дослідженнях ІІІ фази побічні реакції на фоні застосування препарату ЗАЛТРАП (ZALTRAP) в підгрупі пацієнтів з легкою нирковою дисфункцією на вихідному рівні (n=352) були порівнянними із такими у пацієнтів без ниркової дисфункції (n=642). Кількість пацієнтів з помірною/тяжкою нирковою дисфункцією на вихідному рівні, що отримали препарат ЗАЛТРАП (ZALTRAP), була обмеженою (n=49). У цих пацієнтів частота виникнення позаниркових побічних явищ в підгрупі пацієнтів з нирковою дисфункцією була зазвичай порівнянною з такою частотою у пацієнтів без ниркової дисфункції, за винятком виявленого збільшення частоти виникнення дегідратації (усіх ступенів тяжкості) на >10% (див. розділ «Особливості застосування»).
Імуногенність. ЗАЛТРАП (ZALTRAP), як і інші білкові лікарські препарати, є потенційно імуногенним.
За сукупними результатами усіх клінічних досліджень в області онкології спостерігається подібна частота випадків вироблення антитіл до лікарського засобу (АЛЗ) з низьким титром (після початку дослідження) за результатами тестів на АЛЗ, як у пацієнтів, які отримували плацебо, так і у тих, хто приймав ЗАЛТРАП (ZALTRAP) (відповідно 3,3% та 3,8%). Імунна відповідь на афліберсепт з високими титрами АЛЗ не була виявлена у жодного з пацієнтів. Позитивний результат тесту з нейтралізацією антитіл мали 17 пацієнтів, які приймали ЗАЛТРАП (ZALTRAP) (1,6%), та 2 пацієнти, які отримували плацебо (0,2%). У базовому дослідженні за участю пацієнтів з МКРР позитивна відповідь в тесті на АЛЗ частіше спостерігалася у пацієнтів, які отримували плацебо/FOLFIRI [18/526 (3,4%)], ніж у пацієнтів, які отримували ЗАЛТРАП (ZALTRAP)/FOLFIRI [8/521 (1,5%)]. Позитивні результати в тесті з нейтралізацією антитіл за результатами базового дослідження МКРР також були вищими у пацієнтів в групі плацебо/FOLFIRI [2/526 (0,38%)], ніж у пацієнтів в групі ЗАЛТРАП (ZALTRAP)/FOLFIRI [1/521 (0,19%)]. Жодного впливу на фармакокінетичний профіль афліберсепту у пацієнтів з позитивним результатом тестів на імуногенність не спостерігалося.
Беручи до уваги подібні результати тестів на АЛЗ у пацієнтів, які отримували плацебо, і у тих, які приймали ЗАЛТРАП (ZALTRAP), можна зробити висновок, що фактична частота виникнення проявів імуногенності до препарату ЗАЛТРАП (ZALTRAP) згідно із результатами цих тестів, ймовірно, є переоціненою.
Дані стосовно імуногенності значною мірою залежать від чутливості та специфічності тесту. Крім того, на отримані дані стосовно частоти позитивних результатів щодо наявності антитіл впливають декілька факторів, в тому числі особливості поводження зі зразками, час відбору зразків, прийом супутніх лікарських засобів та характер основного захворювання. У зв’язку з цим порівняння частоти випадків утворення антитіл до препарату ЗАЛТРАП (ZALTRAP) з частотою випадків утворення антитіл до інших лікарських засобів може бути некоректним.
Протипоказання
Гіперчутливість до афліберсепту або до будь-якої з допоміжних речовин.
Офтальмологічне/інтравітреальне застосування препарату ЗАЛТРАП (ZALTRAP) з огляду на його гіперосмотичність (див. розділ «Особливості застосування»).
Протипоказання, що стосуються компонентів схеми FOLFIRI (іринотекану, 5-ФУ та фолінової кислоти), див. у відповідних чинних Загальних характеристиках лікарських засобів.
Особливості застосування
Геморагічні явища. У пацієнтів, які отримують афліберсепт, підвищується ризик виникнення геморагічних явищ, в тому числі тяжких і іноді фатальних (див. розділ «Побічні реакції»).
Необхідно моніторувати стан пацієнта для виявлення ознак та симптомів кровотечі з ШКТ, а також інших тяжких геморагічних явищ. Афліберсепт не можна призначати пацієнтам з тяжкими геморагічними явищами (див. розділ «Спосіб застосування та дози»).
У пацієнтів, що отримували лікування засобами ЗАЛТРАП (ZALTRAP)/FOLFIRI, повідомлялося про випадки розвитку тромбоцитопенії. Контроль показників загального аналізу крові з визначенням вмісту тромбоцитів рекомендується на вихідному рівні, перед початком кожного циклу лікування афліберсептом та за клінічними показаннями. Застосування засобів ЗАЛТРАП (ZALTRAP)/FOLFIRI слід тимчасово припинити, поки кількість тромбоцитів не становитиме ≥75 × 109/л (див. розділ «Спосіб застосування та дози»).
Перфорація органів шлунково-кишкового тракту. Повідомлялося про випадки виникнення перфорації органів ШКТ, в тому числі фатальної перфорації органів ШКТ, у пацієнтів, які приймали афліберсепт (див. розділ «Побічні реакції»).
Необхідно моніторувати стан пацієнтів для виявлення ознак та симптомів перфорації органів ШКТ. У випадку виникнення у пацієнта перфорації органів ШКТ застосування афліберсепту слід припинити (див. розділ «Спосіб застосування та дози»).
Утворення нориць. У пацієнтів, які отримували афліберсепт, спостерігалися випадки утворення нориць, як в ШКТ, так і поза ШКТ (див. розділ «Побічні реакції»).
У випадку виникнення у пацієнта нориці застосування афліберсепту слід припинити (див. розділ «Спосіб застосування та дози»).
Артеріальна гіпертензія. У пацієнтів, які отримували засоби ЗАЛТРАП (ZALTRAP)/FOLFIRI, спостерігався підвищений ризик артеріальної гіпертензії 3-4 ступеня тяжкості (в тому числі артеріальна гіпертензія та один випадок есенціальної гіпертензії) (див. розділ «Побічні реакції»).
При існуючій артеріальній гіпертензії необхідно досягти її належного контролю, перш ніж починати лікування афліберсептом. Якщо досягти належного контролю артеріальної гіпертензії не вдається, лікування афліберсептом розпочинати не можна. Під час курсу лікування афліберсептом рекомендується виконувати вимірювання артеріального тиску кожні два тижні, в тому числі перед кожним введенням препарату, або згідно із клінічними показаннями. При виникненні артеріальної гіпертензії на фоні лікування афліберсептом слід забезпечити контроль артеріального тиску за допомогою відповідної антигіпертензивної терапії та регулярно проводити вимірювання артеріального тиску. У випадку тяжкої артеріальної гіпертензії застосування афліберсепту слід тимчасово припинити, поки не буде досягнутий контроль над гіпертензією, і в наступних циклах хіміотерапії зменшити дозу препарату до 2 мг/кг. У випадку неможливості належним чином контролювати артеріальну гіпертензію відповідною антигіпертензивною терапією та у випадку виникнення гіпертензивного кризу або гіпертензивної енцефалопатії афліберсепт слід відмінити остаточно (див. розділ «Спосіб застосування та дози»).
Артеріальна гіпертензія може загострювати перебіг існуючих серцево-судинних захворювань. У пацієнтів з клінічно значущими серцево-судинними захворюваннями у анамнезі, такими як ішемічна хвороба серця або застійна серцева недостатність, лікування препаратом ЗАЛТРАП (ZALTRAP) має здійснюватися з обережністю. Пацієнтам з застійною серцевою недостатністю III або IV класу за NYHA препарат ЗАЛТРАП (ZALTRAP) призначати не можна.
Тромботичні та емболічні явища.
Артеріальні тромбоемболічні події (АТЕ). У пацієнтів, які отримували афліберсепт, спостерігалися випадки АТЕ (в тому числі транзиторна ішемічна атака, цереброваскулярні події, стенокардія, внутрішньосерцевий тромб, інфаркт міокарда, артеріальні емболії та ішемічний коліт) (див. розділ «Побічні реакції»).
У випадку виникнення у пацієнта АТЕ застосування афліберсепту слід припинити (див. розділ «Спосіб застосування та дози»).
Венозні тромбоемболічні події (ВТЕ). У пацієнтів, які отримували афліберсепт, спостерігалися випадки ВТЕ, в тому числі тромбоз глибоких вен (ТГВ) і тромбоемболія легеневої артерії (фатальна у нечастих випадках) (див. розділ «Побічні реакції»).
У пацієнтів з небезпечними для життя (4 ступеня тяжкості) тромбоемболічними явищами (в тому числі тромбоемболією легеневої артерії) застосування препарату ЗАЛТРАП (ZALTRAP) необхідно відмінити (див. розділ «Спосіб застосування та дози»). У пацієнтів з ТГВ 3 ступеня тяжкості слід призначити антикоагулянти за клінічними показаннями та продовжити терапію афліберсептом. У випадку повторного виникнення цих явищ, незважаючи на відповідну антикоагулянтну терапію, застосування афліберсепту слід припинити. У пацієнтів з тромбоемболічними явищами 3 ступеня тяжкості або нижче показане ретельне клінічне моніторування.
Протеїнурія. У пацієнтів, які отримували афліберсепт, спостерігалися тяжка протеїнурія, нефротичний синдром та тромботична мікроангіопатія (ТМА) (див. розділ «Побічні реакції»).
Перед кожним введенням афліберсепту слід контролювати протеїнурію шляхом аналізу сечі за допомогою індикаторної смужки та визначення співвідношення білка і креатиніну в сечі для виявлення розвитку або прогресування протеїнурії. У пацієнтів зі співвідношенням білок/креатинін в сечі >1 слід виконати аналіз сечі, зібраної впродовж 24 годин.
Застосування афліберсепту слід тимчасово припинити при рівні протеїнурії ≥2 г/24 год і відновити, коли рівень протеїнурії становитиме <2 г/24 год. При повторних проявах протеїнурії застосування афліберсепту слід тимчасово припинити до досягнення рівня протеїнурії <2 г/24 год, після чого застосування засобу можна поновити, зменшивши дозу до 2 мг/кг. Застосування афліберсепту слід припинити у пацієнтів, у яких розвинувся нефротичний синдром або ТМА (див. розділ «Спосіб застосування та дози»).
Нейтропенія та нейтропенічні ускладнення. Повідомлялося про підвищення частоти виникнення нейтропенічних ускладнень (фебрильна нейтропенія та нейтропенічна інфекція) при застосуванні комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI (див. розділ «Побічні реакції»).
Контроль показників загального аналізу крові з визначенням лейкоцитарної формули рекомендується на вихідному рівні та перед початком кожного циклу лікування афліберсептом. Застосування засобів ЗАЛТРАП (ZALTRAP)/FOLFIRI слід тимчасово припинити, поки кількість нейтрофілів не становитиме ≥1,5 × 109/л (див. розділ «Спосіб застосування та дози»). У пацієнтів з підвищеним ризиком розвитку нейтропенічних ускладнень може розглядатися можливість терапевтичного застосування Г-КСФ при першому виникненні нейтропенії 3 ступеня тяжкості та вище, а також для вторинної профілактики.
Діарея та дегідратація. Повідомлялося про підвищення частоти виникнення тяжкої діареї при застосуванні комбінації ЗАЛТРАП (ZALTRAP)/FOLFIRI (див. розділ «Побічні реакції»).
За необхідності слід зробити корекцію доз препаратів схеми FOLFIRI (див. розділ «Спосіб застосування та дози»), призначити протидіарейні лікарські засоби та виконати регідратацію.
Реакції гіперчутливості. При проведенні базового дослідження за участю пацієнтів з МКРР у учасників дослідження, які отримували комбінацію ЗАЛТРАП (ZALTRAP)/FOLFIRI, були зареєстровані випадки тяжких реакцій гіперчутливості (див. розділ «Спосіб застосування та дози»).
У випадку виникнення тяжких реакцій гіперчутливості (в тому числі бронхоспазму, задишки, ангіоневротичного набряку та анафілаксії) слід припинити застосування афліберсепту та вжити відповідних медичних заходів (див. розділ «Спосіб застосування та дози»).
При виникненні легких або середньо тяжких реакцій гіперчутливості до препарату ЗАЛТРАП (ZALTRAP) (в тому числі гіперемія, висипання, кропивниця та свербіння) застосування афліберсепту слід тимчасово припинити до зникнення реакції. За наявності клінічних показань можна призначати кортикостероїди та/або антигістамінні засоби. У наступних циклах хіміотерапії може бути розглянута можливість попереднього застосування кортикостероїдів та/або антигістамінних засобів (див. розділ «Спосіб застосування та дози»). При повторному призначенні препарату у пацієнтів, у яких раніше спостерігалися реакції гіперчутливості, необхідно дотримуватися обережності, оскільки у деяких пацієнтів спостерігалися випадки повторного розвитку реакцій гіперчутливості, незважаючи на профілактичні заходи, в тому числі застосування кортикостероїдів.
Порушення загоєння ран. Афліберсепт погіршував загоєння ран в експериментальних моделях на тваринах.
При застосуванні афліберсепту був відмічений потенціал погіршення процесу загоєння ран (розходження країв рани, неспроможність анастомозу) (див. розділ «Побічні реакції»).
Застосування афліберсепту слід припиняти щонайменше за 4 тижні до планових хірургічних втручань.
Застосування афліберсепту рекомендується розпочинати не раніше ніж через 4 тижні після великого хірургічного втручання і лише після повного загоєння операційної рани. Після малого хірургічного втручання, такого як катетеризація центральних вен, біопсія та екстракція зуба, застосування афліберсепту можна розпочинати/поновлювати після повного загоєння операційної рани. У пацієнтів з порушенням загоєння ран, яке потребує медичного втручання, афліберсепт слід відмінити (див. розділ «Спосіб застосування та дози»).
Синдром зворотної задньої енцефалопатії (СЗЗЕ). У базовому дослідженні III фази за участю пацієнтів з МКРР випадків виникнення СЗЗЕ зареєстровано не було. У інших дослідженнях повідомлялося про випадки виникнення СЗЗЕ у пацієнтів, які отримували афліберсепт як у якості монотерапії, так і в комбінації з іншими хіміотерапевтичними засобами (див. розділ «Побічні реакції»).
СЗЗЕ може проявлятися зміненим станом свідомості, судомами, нудотою, блюванням, головним болем або розладами зору. Діагноз СЗЗЕ підтверджується даними магнітно-резонансної томографії (МРТ).
У випадку виникнення у пацієнта СЗЗЕ застосування афліберсепту слід припинити (див. розділ «Спосіб застосування та дози»).
Люди похилого віку. У пацієнтів віком ≥65 років спостерігався підвищений ризик розвитку діареї, запаморочення, загальної слабкості, зниження маси тіла та дегідратації. Рекомендований ретельний нагляд за такими пацієнтами з метою швидкого виявлення ознак та симптомів діареї і дегідратації, призначення відповідного лікування та мінімізації потенційного ризику (див. розділ «Побічні реакції»).
Ниркова дисфункція. Дані щодо лікування афліберсептом пацієнтів з тяжкою дисфункцією нирок наразі дуже обмежені.
Коригування дози афліберсепту не потрібне (див. розділи «Спосіб застосування та дози», «Побічні реакції» та «Фармакокінетичні властивості»).
Функціональний статус та супутні захворювання. Пацієнти з функціональним статусом ≥2 балів за шкалою ECOG або зі значущими супутніми захворюваннями можуть мати підвищений ризик несприятливих клінічних наслідків та повинні підлягати ретельному моніторуванню на предмет виявлення ранніх ознак погіршення клінічного стану.
Інтравітреальне застосування поза схваленими показаннями. ЗАЛТРАП (ZALTRAP) є гіперосмотичним розчином, що був розроблений без огляду на сумісність із внутрішньоочними середовищами. ЗАЛТРАП (ZALTRAP) не можна вводити інтравітреально (див. розділ «Протипоказання»).
Застосування у період вагітності та годування груддю.
Жінки з репродуктивним потенціалом / Контрацепція у чоловіків та жінок. Слід рекомендувати жінкам з репродуктивним потенціалом уникати вагітності в період застосування препарату ЗАЛТРАП (ZALTRAP) та проінформувати їх про потенційну небезпеку цього засобу для плода. Жінки і чоловіки з репродуктивним потенціалом повинні використовувати ефективні контрацептивні засоби під час застосування препарату та протягом щонайменше 6 місяців після прийому останньої дози препарату.
Вагітність. На сьогоднішній день відсутні дані щодо застосування афліберсепту у вагітних жінок. За результатами досліджень на тваринах було показано токсичність препарату для репродуктивної функції. Оскільки ангіогенез має критичне значення для розвитку плода, інгібування ангіогенезу, яке спостерігається при застосуванні препарату ЗАЛТРАП (ZALTRAP), може викликати небажані явища під час вагітності. Призначати ЗАЛТРАП (ZALTRAP) під час вагітності слід тільки у випадках, коли потенційна користь лікування виправдовує потенційний ризик для плода. Якщо пацієнтка завагітніла під час лікування препаратом ЗАЛТРАП (ZALTRAP), її необхідно проінформувати про потенційну небезпеку для плода.
Годування груддю. Жодних досліджень з вивчення впливу препарату ЗАЛТРАП (ZALTRAP) на вироблення грудного молока, наявність препарату в грудному молоці та його впливу на немовля, що вигодовують груддю, не проводилося.
Невідомо, чи екскретується афліберсепт у грудне молоко людини. Ризик для новонароджених/немовлят виключити не можна. Необхідно прийняти рішення щодо доцільності припинення вигодовування дитини грудним молоком або повної/тимчасової відміни препарату ЗАЛТРАП (ZALTRAP), беручи до уваги переваги вигодовування грудним молоком для дитини та переваги терапії цим препаратом для жінки.
Фертильність. Дані досліджень на мавпах дають підстави очікувати, що під час застосування афліберсепту фертильність чоловіків і жінок може порушуватися.
Вплив на здатність керувати транспортними засобами та працювати з механізмами.
Вплив препарату ЗАЛТРАП (ZALTRAP) на здатність керувати транспортними засобами чи працювати з механізмами відсутній чи незначний. Пацієнтам, у яких виникають симптоми, пов'язані з погіршенням зору або зниженням здатності до концентрації уваги чи швидкості реакції, слід рекомендувати відмовитися на період лікування від управління транспортними засобами та роботи з механізмами (див. розділ «Побічні реакції»).
Взаємодія з іншими лікарськими засобами та інші форми взаємодії
Популяційний фармакокінетичний аналіз та порівняння даних різних досліджень між собою не виявили жодних фармакокінетичних лікарських взаємодій між афліберсептом та препаратами схеми FOLFIRI.
Умови зберігання
Зберігати у холодильнику при температурі від +2° С до + 8° С. Зберігати в оригінальній упаковці, щоб захистити препарат від дії світла.
Термін придатності
Закритий флакон: 3 роки.
Після розведення в інфузійному пакеті: хімічна та фізична стабільність готового до використання розчину зберігається протягом 24 годин при температурі від +2° С до + 8° С та 8 годин при температурі + 25°C.
З мікробіологічної точки зору інфузійний розчин повинен бути використаний негайно. Якщо він не використовується одразу ж, за строки та умови його зберігання до початку використання відповідає користувач; зазвичай вони не повинні перевищувати 24 годин при температурі від +2° С до + 8° С, за винятком випадків, коли розведення препарату виконувалося у контрольованих та валідованих асептичних умовах.
Умови відпуску
За рецептом лікаря.
Пакування
Флакон з прозорого боросилікатного скла гідролітичного типу I, закритий ковпачком золотистого кольору зі знімною кришечкою із вставленим ущільнюючим диском, з покриттям Flurotec ® (ПТФЕ).
Флакон, який містить 100 мг / 4,0 мл препарату, має об’єм 5 мл та синю знімну кришечку.
Флакон, який містить 200 мг / 8,0 мл препарату, має об’єм 10 мл та оранжеву знімну кришечку.
По 1 флакону в картонній коробці разом з інструкцією про застосування медичного імунобіологічного препарату.
Виробник
Sanofi-Aventis Deutschland GmbH/ Санофі-Авентіс Дойчланд ГмбХ.
Адреса: Bruningstrasse 50 Н600, H500, H590 65926 Frankfurt am Main, Germany/ Брюнінгштрассе 50 Н600, H500, H590 65926 Франкфурт-на-Майні, Німеччина.
У випадку побічної дії (ускладнення) після застосування МІБП необхідно направити термінове повідомлення до:
Управління лікарських засобів та медичної продукції Міністерства охорони здоров’я України (01021, м. Київ, вул. Грушевського,7, тел: (044) 200-07-93);
Державного підприємства «Державний експертний центр Міністерства охорони здоров’я України» (03151, м. Київ, вул. Ушинського, 40, тел.: (044) 393-75-86);
ТОВ «Санофі-Авентіс Україна», 48-50А, вул. Жилянська, Київ 01033, Україна.
 Торемифен — препарат выбора при блокировании рецепторного звена в антиэстрогенной терапии рака молочной железы
Торемифен — препарат выбора при блокировании рецепторного звена в антиэстрогенной терапии рака молочной железы
 Терапия хронического болевого синдрома в онкологии методом химического нейролизиса
Терапия хронического болевого синдрома в онкологии методом химического нейролизиса
 Как уберечь себя от рака молочной железы
Как уберечь себя от рака молочной железы
 Таргетная терапия: продление и улучшение качества жизни у больных раком молочной железы
Таргетная терапия: продление и улучшение качества жизни у больных раком молочной железы